Peak-to-Gene Linkage#
epione.tl.peak_to_gene ports ArchR’s addPeak2GeneLinks: kNN metacells over a joint embedding → mean-aggregate peak and gene matrices → per-pair Pearson correlation with Student-t + BH FDR.
epione.pl.plot_peak2gene renders an ArchR-style browser track — per-group pseudobulk coverage, peaks, half-ellipse link arcs, and a UCSC-style gene model — without requiring BigWig files.
Data Preparation#
snapatac2 bundles pre-processed h5ad files of the 10x PBMC 10k multiome dataset. ATAC and RNA share barcodes — keep the intersection.
%matplotlib inline
%load_ext autoreload
%autoreload 2
import os, pathlib
os.environ['XDG_CACHE_HOME'] = '/scratch/users/steorra/cache'
import numpy as np
import pandas as pd
import anndata as ad
import scanpy as sc
from IPython.display import display
import epione as epi
import matplotlib.pyplot as plt
epi.pl.plot_set()
WORK = pathlib.Path('/scratch/users/steorra/data/pbmc10k_p2g')
WORK.mkdir(parents=True, exist_ok=True)
└─ 🔬 Starting plot initialization...
├─ Apply Scanpy/matplotlib settings
├─ Custom font setup
├─ Suppress warnings
├─
___________ .__
\_ _____/_____ |__| ____ ____ ____
| __)_\____ \| |/ _ \ / \_/ __ \
| \ |_> > ( <_> ) | \ ___/
/_______ / __/|__|\____/|___| /\___ >
\/|__| \/ \/
├─ 🔖 Version: 0.0.1rc1 📚 Tutorials: https://epione.readthedocs.io/
└─ ✅ plot_set complete.
# Load the PBMC 10k multiome ATAC + RNA h5ad files. Both are in
# the epione dataset registry; pooch downloads + caches them on
# first use.
DS = epi.utils.register_datasets()
atac = ad.read_h5ad(DS.fetch('10x-Multiome-Pbmc10k-ATAC.h5ad'))
rna = ad.read_h5ad(DS.fetch('10x-Multiome-Pbmc10k-RNA.h5ad'))
print(atac)
print(rna)
AnnData object with n_obs × n_vars = 9631 × 107194
obs: 'domain', 'cell_type'
var: 'feature_types'
uns: 'spectral_eigenvalue'
obsm: 'X_spectral', 'X_umap'
AnnData object with n_obs × n_vars = 9631 × 29095
obs: 'domain', 'cell_type'
var: 'gene_ids', 'feature_types'
common = atac.obs_names.intersection(rna.obs_names)
atac = atac[common].copy()
rna = rna[common].copy()
atac.obs['n_fragment'] = np.asarray(atac.X.sum(axis=1)).ravel()
atac.shape, rna.shape
((9631, 107194), (9631, 29095))
Iterative LSI on the ATAC matrix#
peak_to_gene needs a dense low-dimensional embedding to build kNN metacells from. Iterative LSI is the canonical choice for scATAC.
%%time
epi.tl.iterative_lsi(
atac,
n_components=30,
iterations=2,
var_features=25_000,
resolution=0.5,
n_neighbors=30,
sample_cells_pre=10_000,
depth_col='n_fragment',
seed=1,
)
atac.obsm['X_iterative_lsi'].shape
└─ [iterative_lsi] Initial feature set: 106,658 / 107,194
└─ [iterative_lsi] Iter 1/2 | fit on 9,631 cells x 106,658 features
computing neighbors
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:36)
running Leiden clustering
finished: found 15 clusters and added
'leiden', the cluster labels (adata.obs, categorical) (0:00:00)
└─ [iterative_lsi] -> 15 clusters; selected 25,000 variable features for next round
└─ [iterative_lsi] Iter 2/2 | fit on 9,631 cells x 25,000 features
└─ [iterative_lsi] Done. Stored embedding (9,631 x 29) in adata.obsm['X_iterative_lsi']
CPU times: user 2min 26s, sys: 5.14 s, total: 2min 31s
Wall time: 1min 33s
(9631, 29)
Gene annotation#
Per-gene coordinates define the ±max_distance window around each TSS. snapatac2 ships a cached GENCODE hg38 GFF3.
import pyranges as pr
gtf_path = epi.utils.genome.hg38.annotation
g = pr.read_gff3(str(gtf_path)).df
gene_ann = pd.DataFrame({
'gene_name': g.loc[g.Feature == 'gene', 'gene_name'].astype(str).values,
'chrom': g.loc[g.Feature == 'gene', 'Chromosome'].astype(str).values,
'start': g.loc[g.Feature == 'gene', 'Start'].astype(int).values,
'end': g.loc[g.Feature == 'gene', 'End'].astype(int).values,
'strand': g.loc[g.Feature == 'gene', 'Strand'].astype(str).values,
}).drop_duplicates('gene_name').reset_index(drop=True)
exon_ann = pd.DataFrame({
'gene_name': g.loc[g.Feature == 'exon', 'gene_name'].astype(str).values,
'chrom': g.loc[g.Feature == 'exon', 'Chromosome'].astype(str).values,
'start': g.loc[g.Feature == 'exon', 'Start'].astype(int).values,
'end': g.loc[g.Feature == 'exon', 'End'].astype(int).values,
'strand': g.loc[g.Feature == 'exon', 'Strand'].astype(str).values,
})
len(gene_ann), len(exon_ann)
(60606, 839796)
Peak-to-gene#
ArchR defaults: 500 metacells × 100 neighbours, ±250 kb window, Pearson correlation, BH FDR across all tested pairs.
%%time
links = epi.tl.peak_to_gene(
atac,
rna=rna,
gene_annotation=gene_ann,
use_rep='X_iterative_lsi',
n_metacells=500,
k_neighbors=100,
max_distance=250_000,
seed=1,
)
links.to_parquet(WORK / 'peak_to_gene.parquet')
print(f'{len(links):,} pairs | {(links.fdr < 0.05).sum():,} FDR < 0.05')
└─ [peak_to_gene] 107,194 peaks | 20,169 annotated genes | 9,631 cells
└─ [peak_to_gene] Building 500 metacells × 100 neighbours from X_iterative_lsi
└─ [peak_to_gene] Aggregating peak matrix
└─ [peak_to_gene] Aggregating gene matrix
└─ [peak_to_gene] Finding peak-gene pairs within ±250,000 bp
└─ [peak_to_gene] 726,292 candidate pairs
└─ [peak_to_gene] Computing correlations
└─ [peak_to_gene] 726,292 pairs retained, 441,142 significant (FDR < 0.05)
726,292 pairs | 441,142 FDR < 0.05
CPU times: user 6.62 s, sys: 1.67 s, total: 8.29 s
Wall time: 7.61 s
links.reindex(links['correlation'].abs().sort_values(ascending=False).index) \
.head(10)[['peak', 'gene', 'distance', 'correlation', 'fdr']]
| peak | gene | distance | correlation | fdr | |
|---|---|---|---|---|---|
| 652165 | chr7:101367780-101367789 | LINC01007 | -201222 | 0.999998 | 0.0 |
| 652164 | chr7:101367780-101367789 | COL26A1 | 4910 | 0.999798 | 0.0 |
| 467543 | chr20:9388473-9388582 | LAMP5-AS1 | -126471 | 0.999493 | 0.0 |
| 467542 | chr20:9388473-9388582 | LAMP5 | -125830 | 0.998663 | 0.0 |
| 360128 | chr19:5336190-5336587 | PTPRS | -4424 | 0.998541 | 0.0 |
| 89849 | chr10:89809057-89809192 | LINC01374 | -44470 | 0.998295 | 0.0 |
| 100520 | chr11:634199-634286 | SCT | 7061 | 0.997344 | 0.0 |
| 679193 | chr8:134625591-134625702 | ZFAT-AS1 | 27576 | 0.997196 | 0.0 |
| 237289 | chr15:75018870-75019357 | SCAMP5 | 61895 | 0.996811 | 0.0 |
| 679199 | chr8:134683132-134683772 | ZFAT-AS1 | 85382 | 0.996300 | 0.0 |
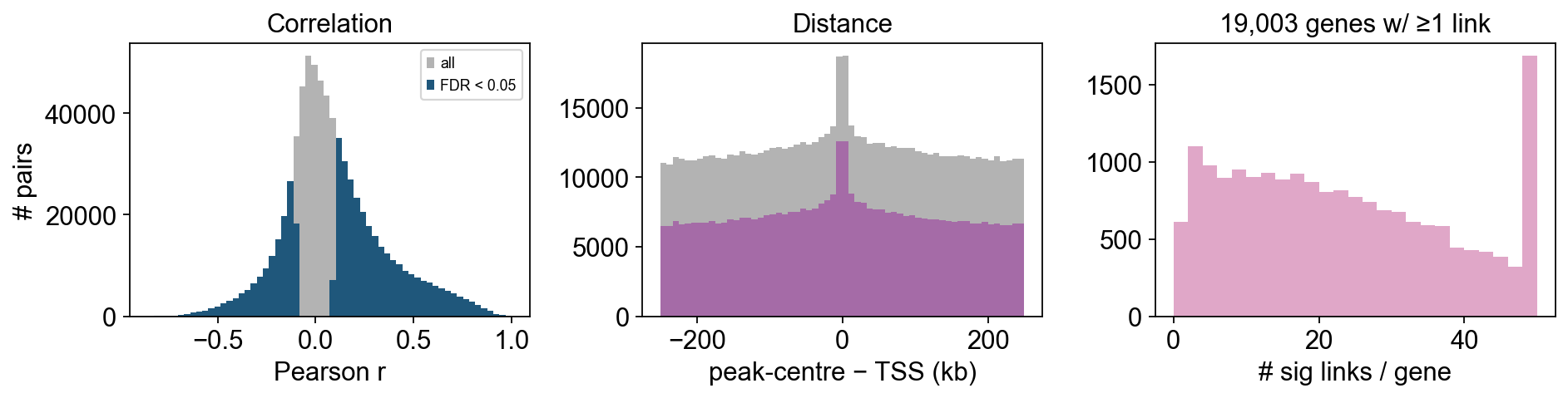
Summary plots#
sig = links[links['fdr'] < 0.05]
fig, axes = plt.subplots(1, 3, figsize=(12, 3.2))
axes[0].hist(links['correlation'], bins=60, color='0.7', label='all')
axes[0].hist(sig['correlation'], bins=60, color='C0', label='FDR < 0.05')
axes[0].set_xlabel('Pearson r'); axes[0].set_ylabel('# pairs')
axes[0].legend(fontsize=8); axes[0].set_title('Correlation')
axes[1].hist(links['distance'] / 1e3, bins=60, color='0.7')
axes[1].hist(sig['distance'] / 1e3, bins=60, color='C1')
axes[1].set_xlabel('peak-centre − TSS (kb)'); axes[1].set_title('Distance')
per_gene = sig.groupby('gene').size().clip(upper=50)
axes[2].hist(per_gene, bins=np.arange(0, 52, 2), color='C2')
axes[2].set_xlabel('# sig links / gene')
axes[2].set_title(f'{per_gene.size:,} genes w/ ≥1 link')
plt.tight_layout(); display(fig); plt.close(fig)
for gene in ['CD3D', 'CD19', 'MS4A1', 'GNLY', 'LYZ']:
sub = sig[sig['gene'] == gene]
if sub.empty:
continue
print(f'\n{gene}: {len(sub)} significant links')
print(sub.reindex(sub.correlation.abs().sort_values(ascending=False).index)
.head(5)[['peak', 'distance', 'correlation']].to_string(index=False))
CD3D: 52 significant links
peak distance correlation
chr11:118337765-118347252 -197 0.882967
chr11:118304165-118308090 -36578 0.849636
chr11:118315296-118317685 -26215 0.779693
chr11:118398435-118402795 57910 0.710349
chr11:118434093-118439206 93944 0.694896
CD19: 44 significant links
peak distance correlation
chr16:28910024-28911110 -21397 0.913139
chr16:28930484-28933437 -4 0.874558
chr16:28923628-28926451 -6925 0.711179
chr16:28902003-28904135 -28895 0.576940
chr16:29007438-29010500 77005 -0.428174
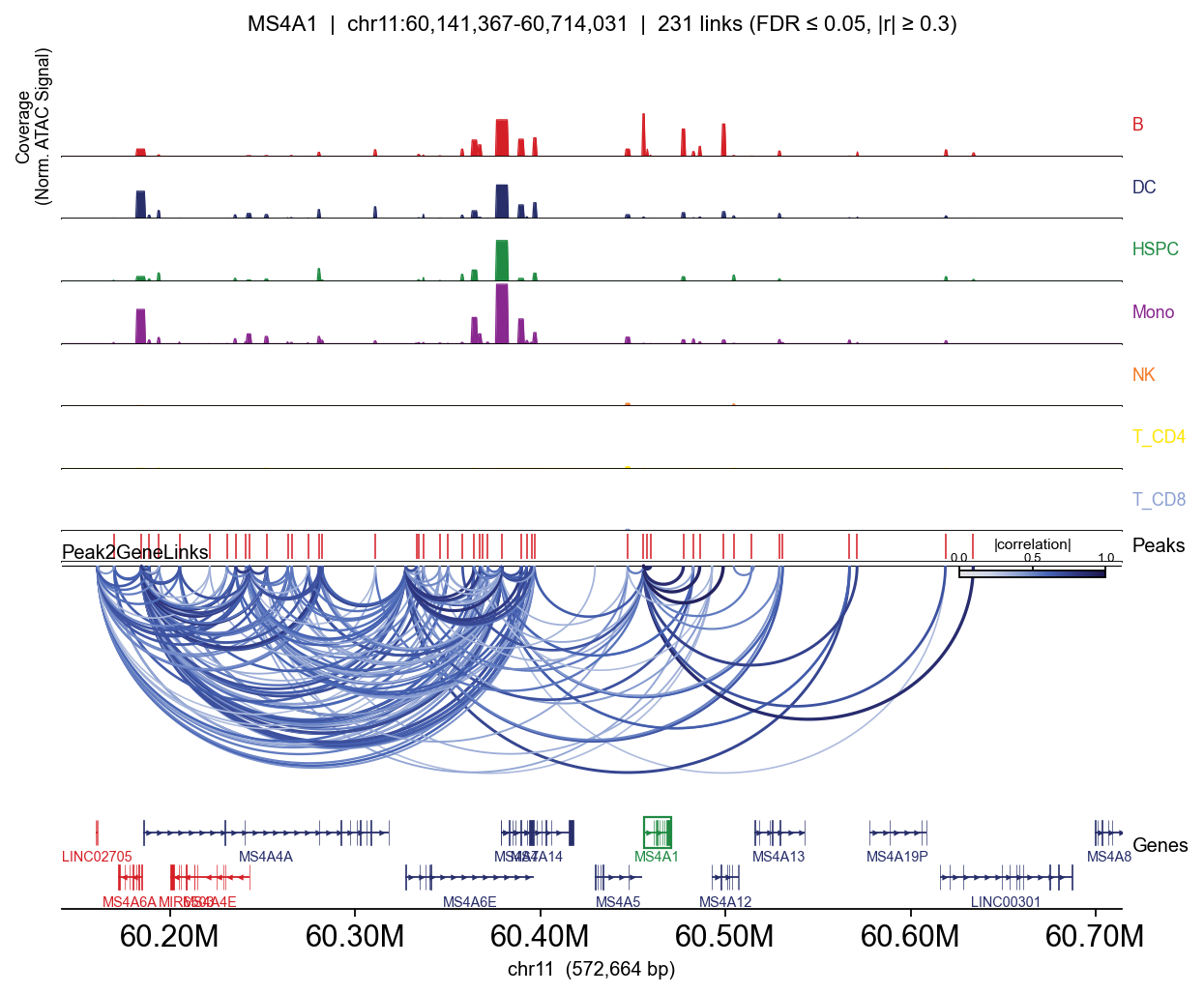
MS4A1: 40 significant links
peak distance correlation
chr11:60455290-60456088 -156 0.958685
chr11:60498212-60499391 42956 0.917259
chr11:60457557-60458006 1936 0.904648
chr11:60485853-60486383 30273 0.900626
chr11:60459521-60459691 3761 0.883234
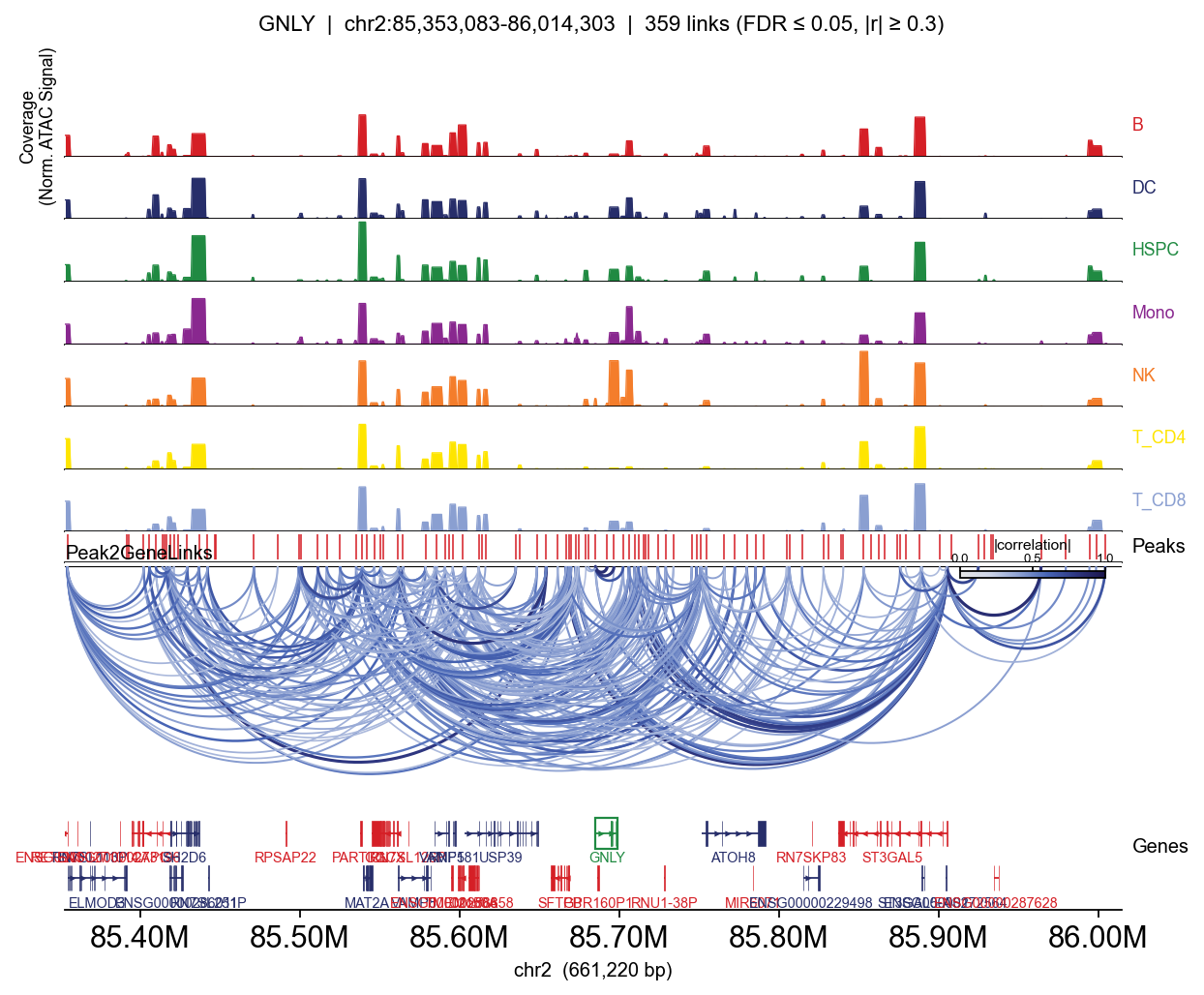
GNLY: 53 significant links
peak distance correlation
chr2:85692097-85692527 7138 0.856677
chr2:85684653-85685264 -216 0.831111
chr2:85693711-85699520 11441 0.806101
chr2:85701405-85703391 17224 0.661557
chr2:85850937-85855115 167852 0.524986
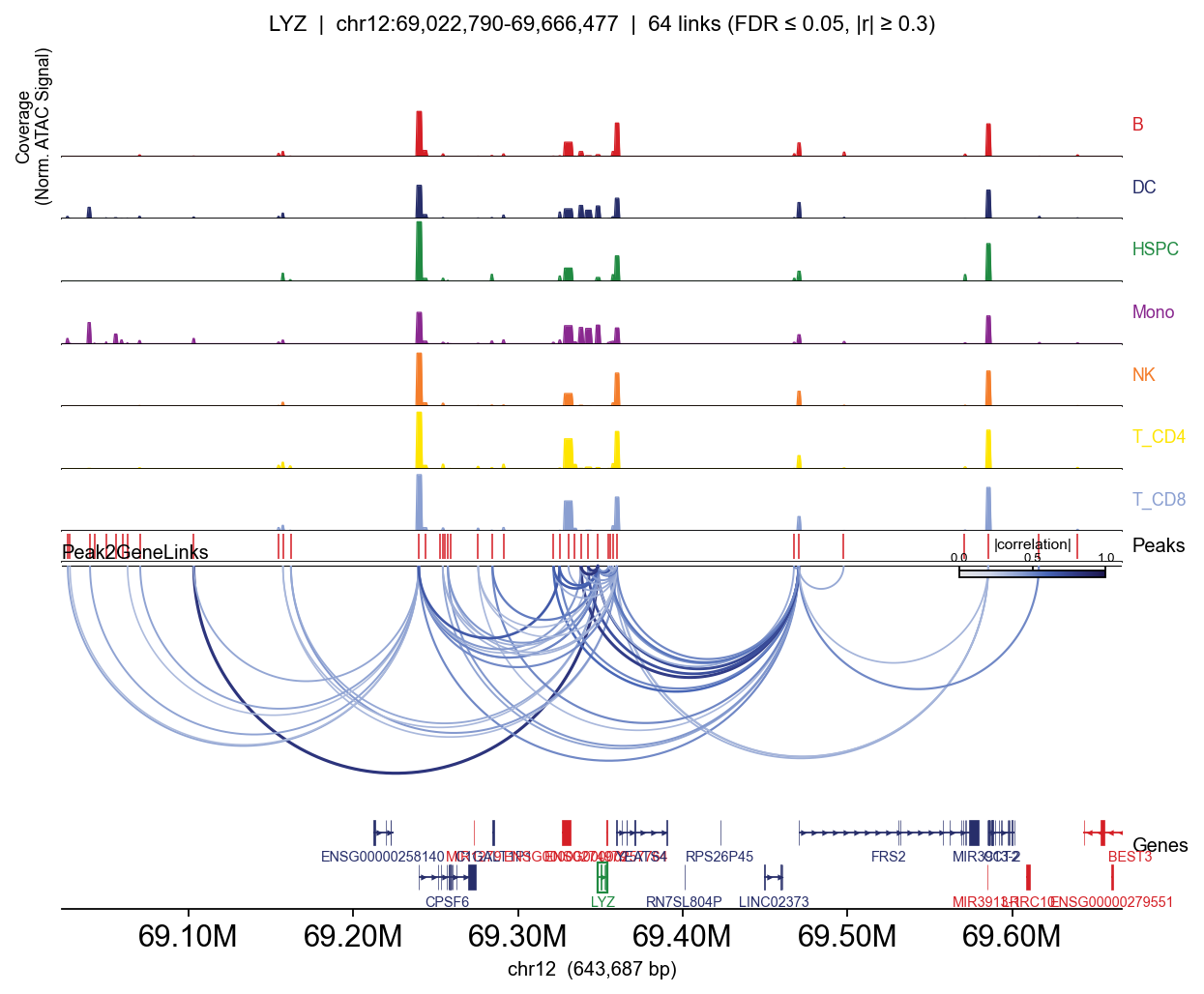
LYZ: 28 significant links
peak distance correlation
chr12:69102790-69103314 -245328 0.820270
chr12:69337416-69339102 -10121 0.807979
chr12:69347540-69349292 36 0.794729
chr12:69341126-69344066 -5784 0.788283
chr12:69355939-69356394 7786 0.671455
Visualise links#
epi.pl.plot_peak2gene draws coverage + peaks + link arcs + gene track on a genomic window around a gene. By default no BigWig files are required — per-group coverage is computed on the fly from the peak matrix with per-cell CP10k normalisation.
# Map fine-grained cell types to broad lineages for clearer coverage
major = {
'CD4 Naive': 'T_CD4', 'CD4 TCM': 'T_CD4', 'CD4 TEM': 'T_CD4', 'Treg': 'T_CD4',
'CD8 Naive': 'T_CD8', 'CD8 TEM_1': 'T_CD8', 'CD8 TEM_2': 'T_CD8',
'MAIT': 'T_CD8', 'gdT': 'T_CD8',
'Naive B': 'B', 'Memory B': 'B', 'Intermediate B': 'B', 'Plasma': 'B',
'NK': 'NK', 'CD14 Mono': 'Mono', 'CD16 Mono': 'Mono',
'cDC': 'DC', 'pDC': 'DC', 'HSPC': 'HSPC',
}
atac.obs['lineage'] = atac.obs['cell_type'].map(major).astype('category')
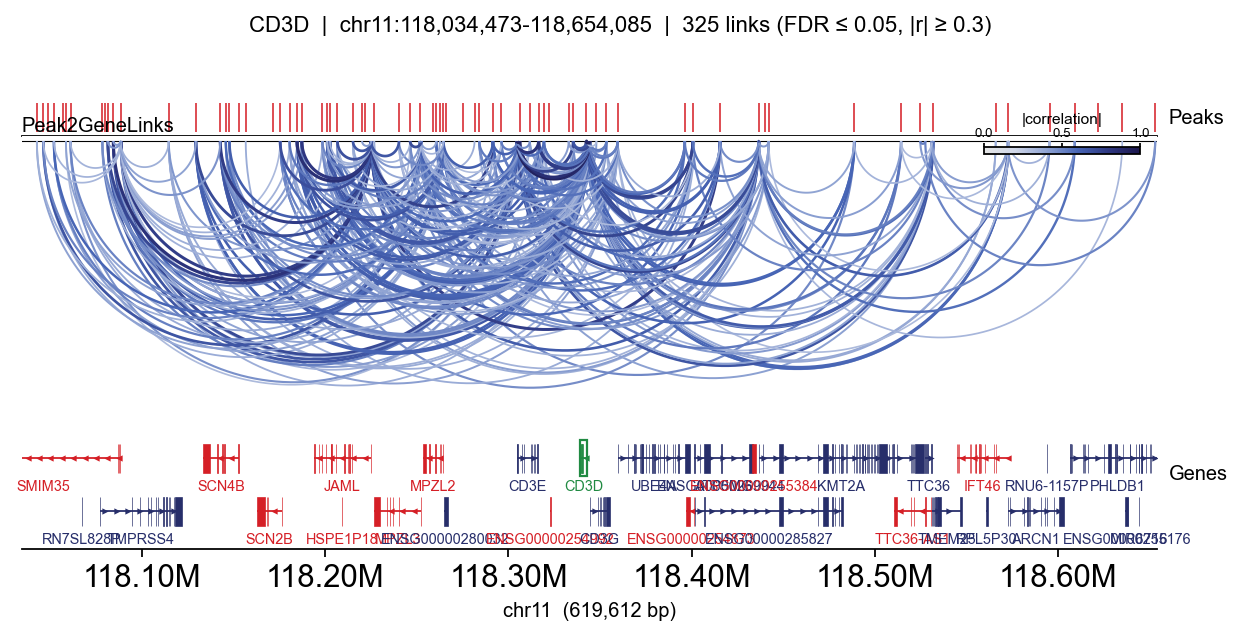
Minimal: arcs + peaks + gene track#
fig, _ = epi.pl.plot_peak2gene(
atac, gene='CD3D',
gene_annotation=gene_ann,
exon_annotation=exon_ann,
fdr_thresh=0.05, min_abs_r=0.3,
pad_bp=80_000, figsize=(8, 4), show=False,
)
plt.tight_layout(); display(fig); plt.close(fig)
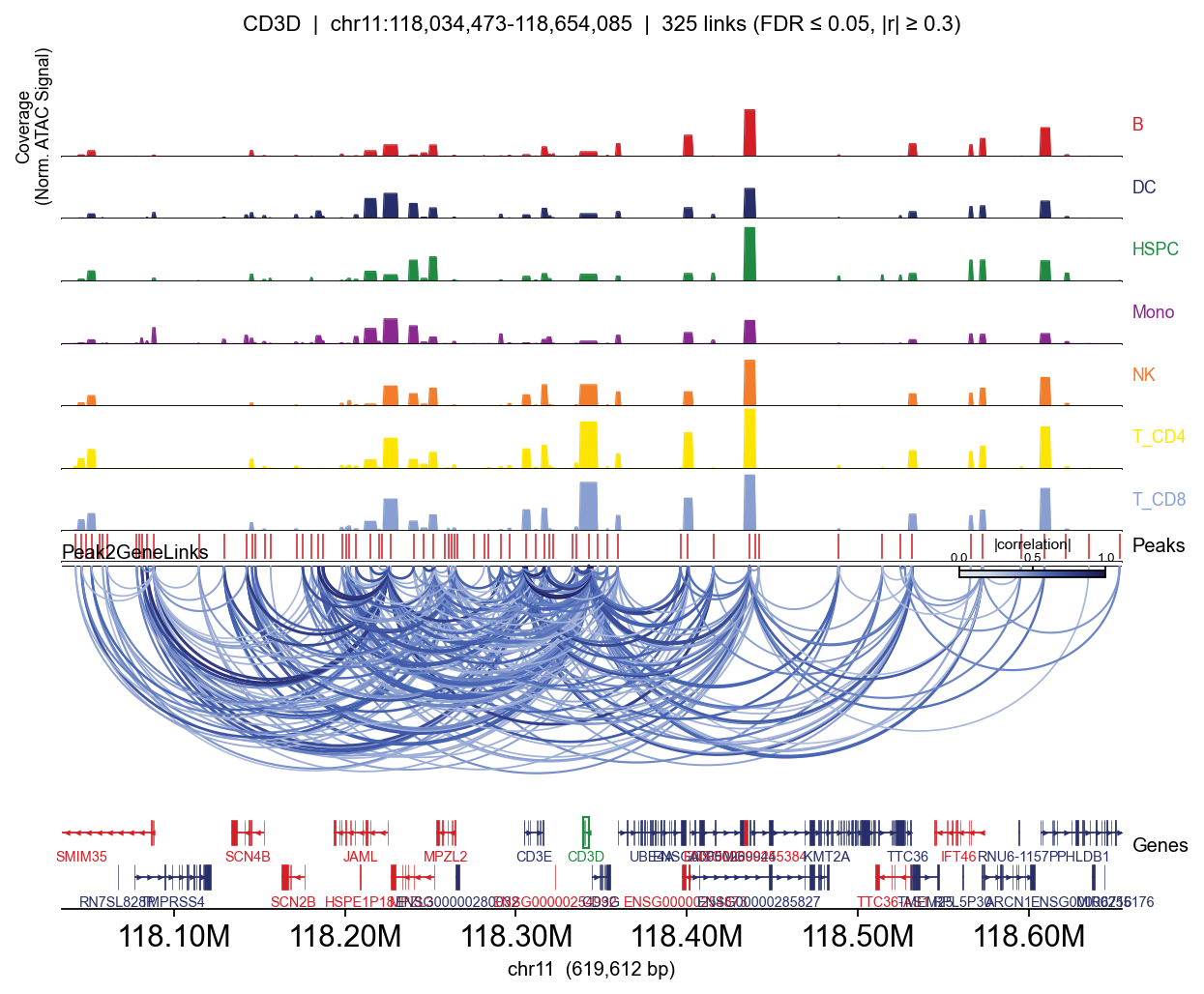
Full track — per-lineage coverage#
Passing group_by= adds one coverage row per category (per-cell CP10k-normalised, mean within group — so T-specific peaks appear clearly in T lineages and not elsewhere).
for gene in ['CD3D', 'MS4A1', 'LYZ', 'GNLY']:
fig, _ = epi.pl.plot_peak2gene(
atac, gene=gene,
group_by='lineage',
gene_annotation=gene_ann,
exon_annotation=exon_ann,
fdr_thresh=0.05, min_abs_r=0.3,
pad_bp=80_000, figsize=(8, 4), show=False,
)
plt.tight_layout(); display(fig); plt.close(fig)
Fragment-based coverage (BigWig pseudobulk)#
The tracks above compute per-lineage coverage from the peak matrix: for each peak, the mean CP10k value within the group is painted across the peak’s width. Fast (no extra I/O), but blocky — the resolution is the peak width, not bp.
For ArchR / UCSC-quality tracks we need bp-resolution Tn5 insertion density, which requires the fragment file. epione.single.pseudobulk_with_fragments does this in one call:
Reads the fragment
.tsv.gz(optionally cached as parquet for re-use).Expands each fragment to two 1-bp Tn5 cutsites with the standard ATAC shift
(+4, -5)— or, withbigwig_strategy='fragment', leaves the fragment interval intact.Writes one CPM-normalised BigWig per group via the Rust-backed
pybigtoolswriter.executor='process'forks workers that share fragments via copy-on-write → true parallel writes (no GIL).
Pass the resulting BWs into plot_peak2gene(bigwig_files=...) and the coverage row swaps to bp-true signal; arcs / peaks / gene rows are unchanged.
frag_path = epi.utils.register_datasets().fetch('pbmc_10k_atac.tsv.gz')
print('fragment file:', frag_path)
fragment file: /scratch/users/steorra/cache/snapatac2/pbmc_10k_atac.tsv.gz
Build per-lineage BigWigs#
Barcodes in snapatac2’s bundled fragment file carry no sample tag — pass sample_id_col=None and a single-entry path_to_fragments. Caching the parsed fragments as parquet pays for itself the second time you tweak parameters.
%%time
BW_DIR = WORK / 'pseudobulk_bw'
BW_DIR.mkdir(exist_ok=True)
chrom = pr.PyRanges(pd.DataFrame({
'Chromosome': list(epi.utils.genome.hg38.chrom_sizes.keys()),
'Start': 0,
'End': list(epi.utils.genome.hg38.chrom_sizes.values()),
}))
epi.single.pseudobulk_with_fragments(
input_data=atac, # AnnData with obs['lineage']
chromsizes=chrom,
cluster_key='lineage',
path_to_fragments={'pbmc10k': str(frag_path)},
bigwig_path=str(BW_DIR),
bed_path=None, # skip BED — slow, rarely needed
bigwig_strategy='cutsite', # Tn5 insertion density (+4/-5)
normalize_bigwig=True, # CPM
cache_fragments=str(WORK / 'frag_cache'),
executor='process', # fork pool for true parallel writes
n_jobs=4,
balance_clusters=False,
show_progress=False,
verbose=False,
)
bigwig_files = {p.stem: str(p) for p in BW_DIR.glob('*.bw')}
bigwig_files
CPU times: user 28.5 s, sys: 14.5 s, total: 43 s
Wall time: 1min 50s
{'T_CD8': '/scratch/users/steorra/data/pbmc10k_p2g/pseudobulk_bw/T_CD8.bw',
'HSPC': '/scratch/users/steorra/data/pbmc10k_p2g/pseudobulk_bw/HSPC.bw',
'NK': '/scratch/users/steorra/data/pbmc10k_p2g/pseudobulk_bw/NK.bw',
'Mono': '/scratch/users/steorra/data/pbmc10k_p2g/pseudobulk_bw/Mono.bw',
'T_CD4': '/scratch/users/steorra/data/pbmc10k_p2g/pseudobulk_bw/T_CD4.bw',
'DC': '/scratch/users/steorra/data/pbmc10k_p2g/pseudobulk_bw/DC.bw',
'B': '/scratch/users/steorra/data/pbmc10k_p2g/pseudobulk_bw/B.bw'}
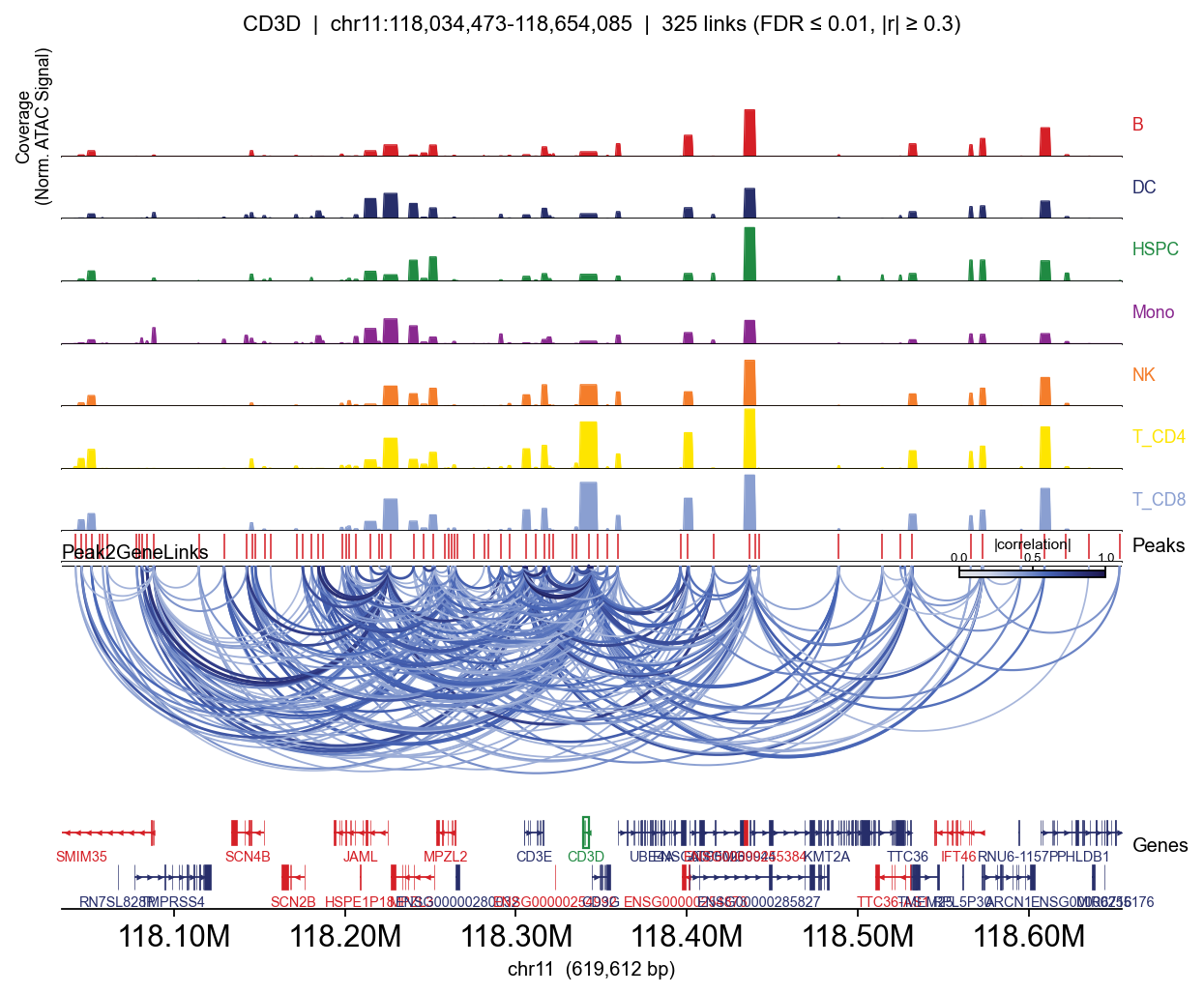
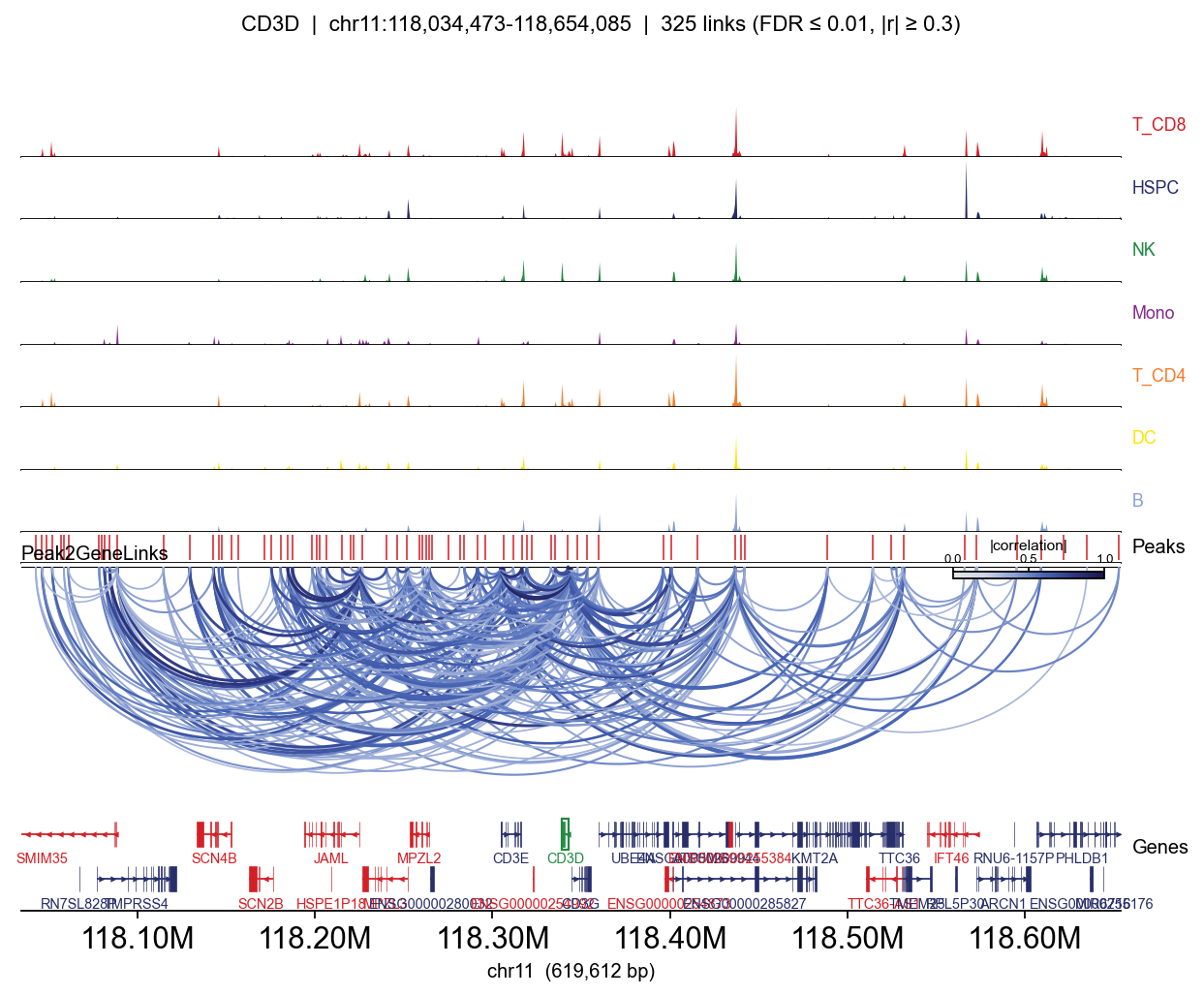
Side-by-side: peak-matrix approximation vs. fragment BigWig#
Same gene (CD3D), same links, same gene track — only the coverage rows differ. The BigWig version shows sharp bp-resolution peaks inside each accessible region; the peak-matrix version paints each peak as a flat block.
for mode, kwargs in [
('peak-matrix (group_by)', dict(group_by='lineage')),
('BigWig (fragments)', dict(bigwig_files=bigwig_files)),
]:
print(f'\n=== {mode} ===')
fig, _ = epi.pl.plot_peak2gene(
atac, gene='CD3D',
gene_annotation=gene_ann,
exon_annotation=exon_ann,
fdr_thresh=0.01, min_abs_r=0.3,
pad_bp=80_000, figsize=(8, 4), show=False,
**kwargs,
)
plt.tight_layout(); display(fig); plt.close(fig)
=== peak-matrix (group_by) ===
=== BigWig (fragments) ===
Notes#
No RNA? Pass a gene-score / gene-activity matrix instead via
gene_obsm='X_gene_score'+gene_names=.... API is otherwise identical.Memory scales with candidate pairs, not cells. 250 kb × 20k genes → ≈20 hits/peak, ~
n_peaks × 20float32.n_metacells=500, k_neighbors=100matches ArchR; bump both for higher resolution at the cost of RAM.Arcs use ArchR’s exact
getArchDFhalf-ellipse geometry (ry = R_MAX · rx / max_rx); gene track follows the UCSC convention (intron line + exon blocks + strand chevrons) whenexon_annotation=is provided.