Hi-C basics — Drosophila S2 contact matrix with epione.bulk.hic / epione.upstream#
End-to-end visual walk-through of epione.bulk.hic / epione.upstream on real Drosophila
Schneider 2 (S2) cell Hi-C — the dataset the Galaxy HiCExplorer
tutorial uses (Ramírez et al. 2018).
We start from the pre-built balanced .cool and show the canonical
Hi-C QC plots: genome-wide heatmap, single-chromosome zoom,
contact-decay P(s) curve, and per-bin coverage + ICE weight diagnostics.
Phase 2 / 3 tutorials (coming next) will build compartments (A/B via
eigendecomposition), TAD boundaries (insulation score), and loop
calls (dot-finder) on top of the same .cool.
Upstream recipe (not run here — ~15 min wall-time)#
The .cool used below was produced from the Galaxy Zenodo dataset
via the 4DN-standard bwa mem -SP5M + pairtools chain:
bwa index dm3_genome.fasta
bwa mem -SP5M -t 8 dm3_genome.fasta R1.fq.gz R2.fq.gz \
| pairtools parse -c chrom.sizes --assembly dm3 --drop-sam --drop-seq --min-mapq 30 \
| pairtools sort --nproc 8 \
| pairtools dedup --mark-dups \
| pairtools select "(pair_type=='UU') or (pair_type=='UR') or (pair_type=='RU')" \
-o drosophila.pairs.gz
cooler cload pairs --assembly dm3 -c1 2 -p1 3 -c2 4 -p2 5 \
chrom.sizes:10000 drosophila.pairs.gz drosophila.cool
In epione.bulk.hic / epione.upstream this maps to the Phase 1 API:
pairs_from_bam(...)— runs the parse/sort/dedup/select chain. (Not invoked here because it needs a real Hi-C BAM; replacingbwawith the existingepi.upstream.bwa_mem2binding is a Phase 2 task.)pairs_to_cool(pairs, sizes, out_cool, binsize=10_000)— load pairs into a.cool.balance_cool(out_cool)— ICE balance in place.
Dataset#
Galaxy HiCExplorer tutorial data-hic-drosophila from
Zenodo record 16416373:
HiC_S2_1p_10min_lowU_R{1,2}.fastq.gz(paired-end Hi-C from Kc167 S2 cells)dm3_genome.fasta— reference genome build dm3
On this run: 16 880 × 10 kb bins, 1.32 M non-zero pixels, 15 chromosomes.
1 · Setup#
import pathlib
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import cooler
import epione as epi
epi.pl.plot_set()
DATA = pathlib.Path('/scratch/users/steorra/data/drosophila-hic')
COOL = DATA / 'drosophila.cool'
assert COOL.exists(), (
f'{COOL} missing — generate it once with the upstream recipe above '
'or point COOL at your own .cool'
)
OUT = pathlib.Path('result') / 't_hic_basics'
OUT.mkdir(parents=True, exist_ok=True)
clr = cooler.Cooler(str(COOL))
print(f'bins : {len(clr.bins()[:]):,}')
print(f'nnz pixels: {clr.pixels()[:].shape[0]:,}')
print(f'binsize : {clr.binsize:,} bp')
print(f'chroms : {list(clr.chromnames)}')
└─ 🔬 Starting plot initialization...
├─ Apply Scanpy/matplotlib settings
├─ Custom font setup
├─ Suppress warnings
├─
___________ .__
\_ _____/_____ |__| ____ ____ ____
| __)_\____ \| |/ _ \ / \_/ __ \
| \ |_> > ( <_> ) | \ ___/
/_______ / __/|__|\____/|___| /\___ >
\/|__| \/ \/
├─ 🔖 Version: 0.0.1rc1 📚 Tutorials: https://epione.readthedocs.io/
└─ ✅ plot_set complete.
bins : 16,880
nnz pixels: 1,317,818
binsize : 10,000 bp
chroms : ['chr2L', 'chr2LHet', 'chr2R', 'chr2RHet', 'chr3L', 'chr3LHet', 'chr3R', 'chr3RHet', 'chr4', 'chrU', 'chrUextra', 'chrX', 'chrXHet', 'chrYHet', 'chrM']
2 · Genome-wide contact matrix#
Every epione.bulk.hic / epione.upstream plotting function reads the .cool directly via
cooler, so to render the whole genome we just pass chrom=None and
tick the chromosome boundaries by hand. For a quick overview we also
sum over chromosomes within a single heatmap to get the cis+trans
contact landscape.
mat_gw = clr.matrix(balance=True)[:]
print('genome-wide matrix:', mat_gw.shape,
'— finite cells:', int(np.isfinite(mat_gw).sum()))
fig, ax = plt.subplots(figsize=(6.5, 6))
from matplotlib.colors import LogNorm
vmax = float(np.nanquantile(mat_gw[np.isfinite(mat_gw)], 0.99))
img = ax.imshow(
np.log10(mat_gw + 1e-5),
origin='upper', cmap='YlOrRd',
vmin=np.log10(max(vmax * 1e-4, 1e-5)), vmax=np.log10(vmax),
interpolation='none',
)
# chromosome tick marks
chrom_edges = clr.offset(clr.chromnames[-1])
offsets = [clr.offset(c) for c in clr.chromnames]
mids = [(o + (offsets[i+1] if i+1 < len(offsets) else clr.shape[0])) / 2
for i, o in enumerate(offsets)]
ax.set_xticks(mids)
ax.set_xticklabels(clr.chromnames, rotation=90, fontsize=7)
ax.set_yticks(mids)
ax.set_yticklabels(clr.chromnames, fontsize=7)
for o in offsets[1:]:
ax.axvline(o, color='k', lw=0.25, alpha=0.5)
ax.axhline(o, color='k', lw=0.25, alpha=0.5)
ax.set_title('Drosophila S2 Hi-C — genome-wide (log contacts, balanced)')
fig.colorbar(img, ax=ax, shrink=0.7, label=r'log$_{10}$ contact')
plt.tight_layout()
plt.show()
genome-wide matrix: (16880, 16880) — finite cells: 164070481
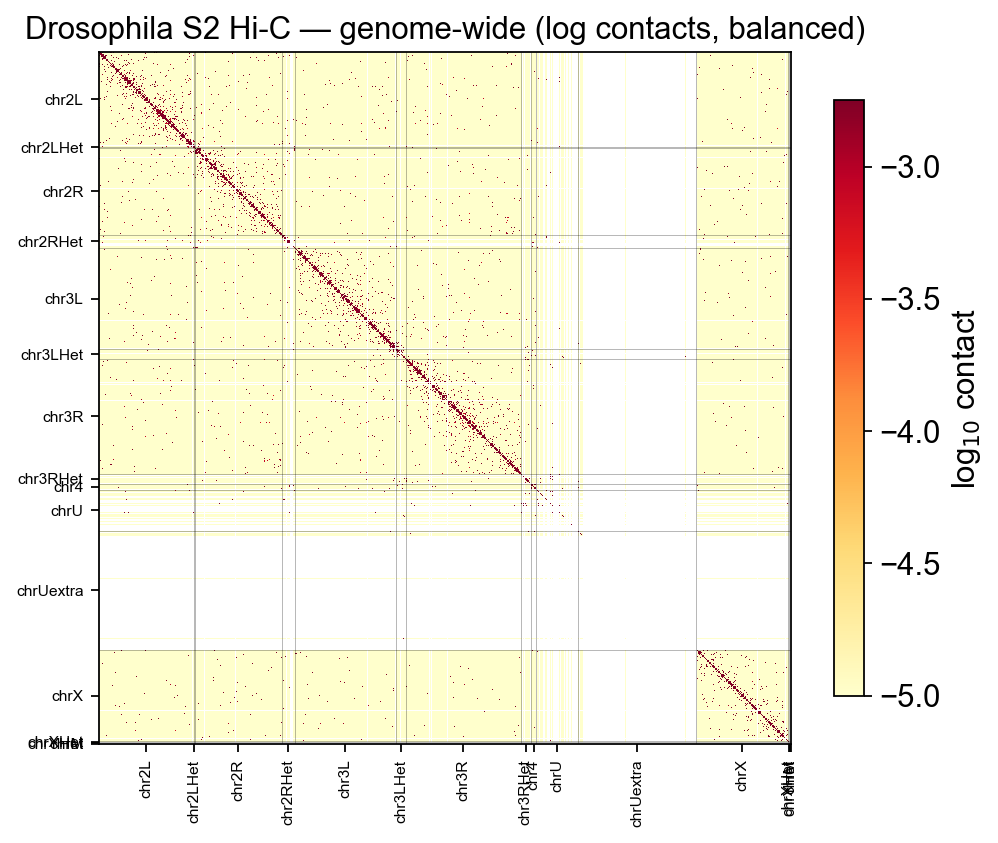
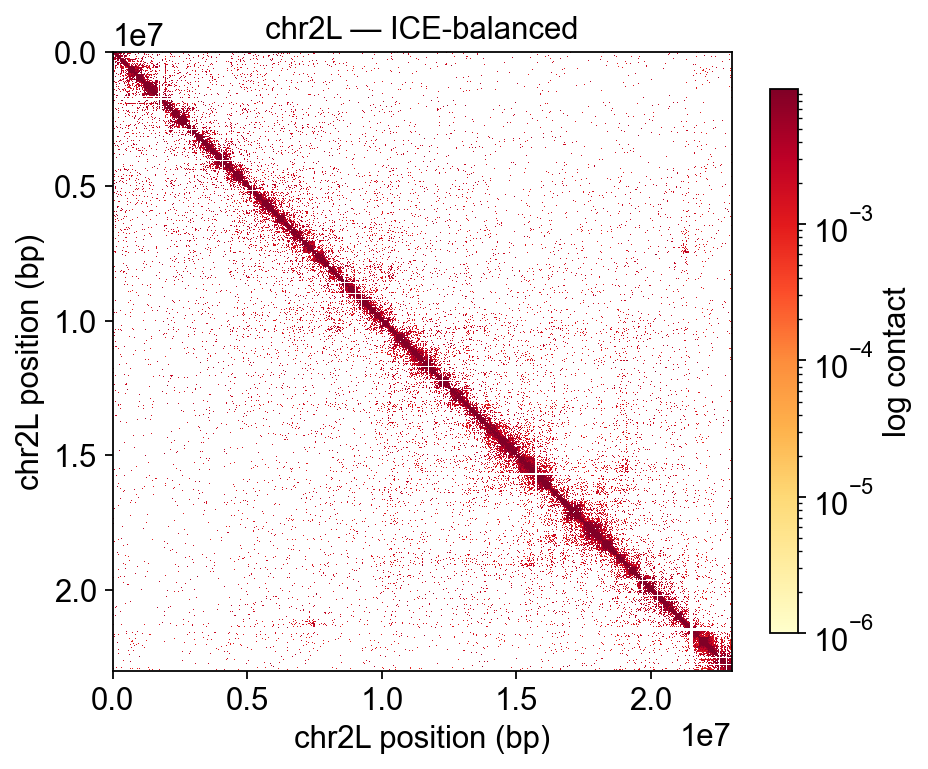
3 · Single-chromosome heatmap#
epi.bulk.hic.plot_contact_matrix takes a UCSC-style region string. The
chr2L 23-Mb arm is the canonical chromosome for tutorials because
it fits comfortably in a single square and has well-studied Polycomb
/ chromatin domains visible as off-diagonal blocks.
fig, ax, img = epi.pl.plot_contact_matrix(
COOL, region='chr2L',
balance=True, log=True,
figsize=(6.2, 5.6),
title='chr2L — ICE-balanced',
)
plt.show()
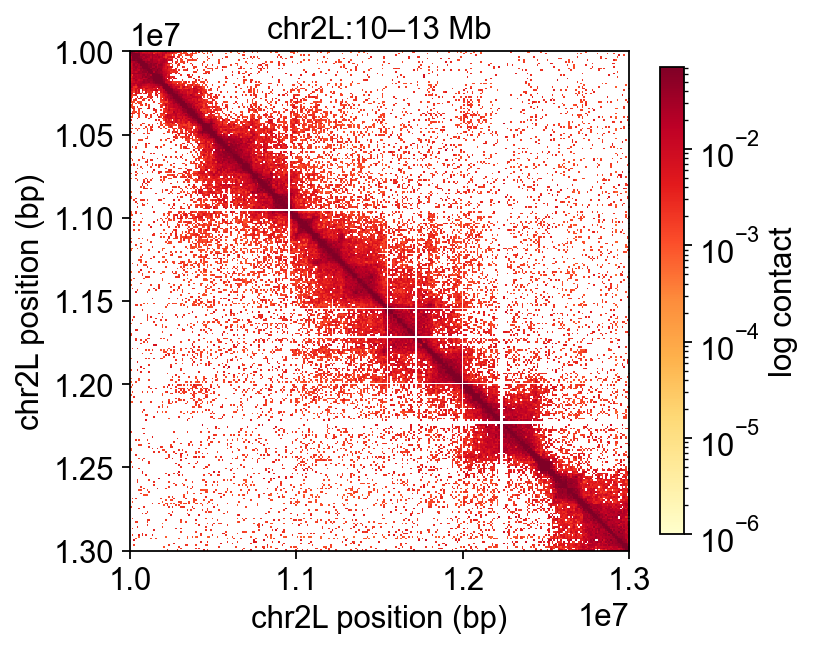
3.1 Zoom into a 3-Mb slice#
Pan into a 3-Mb interval on chr2L so the off-diagonal “boxes” characteristic of Drosophila TADs become visible. Phase 3 will auto-detect the boundaries with insulation score; here we just eye them.
fig, ax, img = epi.pl.plot_contact_matrix(
COOL, region='chr2L:10_000_000-13_000_000',
balance=True, log=True,
figsize=(5.0, 4.8),
title='chr2L:10–13 Mb',
)
plt.show()
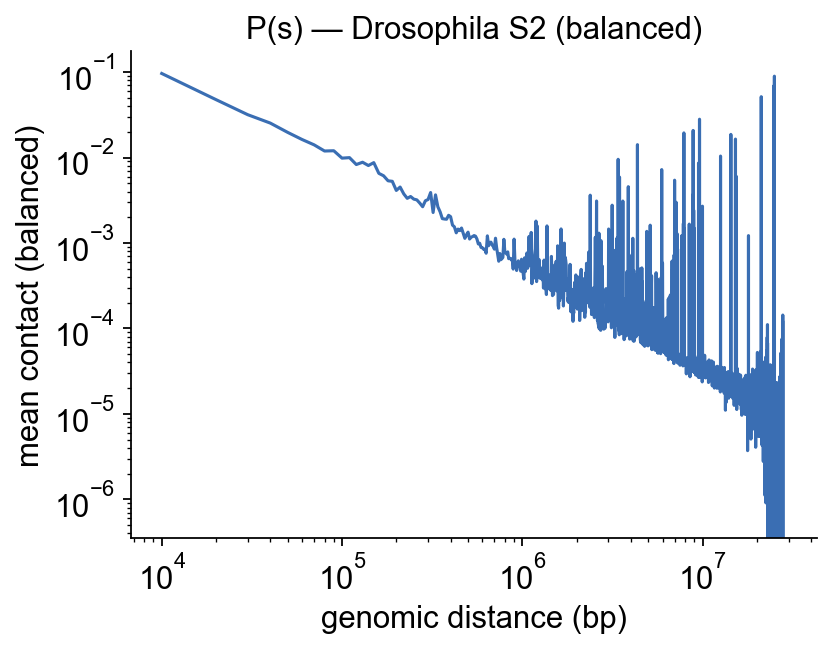
4 · Contact-decay curve P(s)#
The most informative per-library QC plot. Average every intra-chrom diagonal offset across all 15 chromosomes, plot on log-log axes. A healthy Hi-C library decays roughly as a power-law with slope ~−1 over most of the dynamic range; a plateau, bump or non-monotone shape points to undersampling, too-short fragment selection, or over-balancing.
fig, ax, decay_df = epi.pl.plot_decay_curve(
COOL, balance=True, figsize=(5.5, 4.0),
title='P(s) — Drosophila S2 (balanced)',
)
plt.show()
decay_df.head(10)
| chrom | offset_bin | distance_bp | mean_contact | n_pairs | |
|---|---|---|---|---|---|
| 0 | chr2L | 0 | 0 | 0.263320 | 2222 |
| 1 | chr2L | 1 | 10000 | 0.120140 | 2177 |
| 2 | chr2L | 2 | 20000 | 0.056927 | 2167 |
| 3 | chr2L | 3 | 30000 | 0.037639 | 2168 |
| 4 | chr2L | 4 | 40000 | 0.028203 | 2164 |
| 5 | chr2L | 5 | 50000 | 0.022670 | 2158 |
| 6 | chr2L | 6 | 60000 | 0.018948 | 2154 |
| 7 | chr2L | 7 | 70000 | 0.016106 | 2149 |
| 8 | chr2L | 8 | 80000 | 0.013840 | 2150 |
| 9 | chr2L | 9 | 90000 | 0.012063 | 2148 |
5 · Per-bin coverage + ICE weight diagnostics#
plot_coverage lays out two panels side-by-side: left = per-bin
total contact coverage (what ICE balance saw), right = per-bin
ICE weight (what balance emitted). Bins whose weight is NaN are
masked from the balanced matrix — typically low-mappability
regions, gaps, and heterochromatin repeats.
fig, axes = epi.pl.plot_coverage(
COOL, balance=False, bins=50, figsize=(9, 3.5),
)
plt.show()
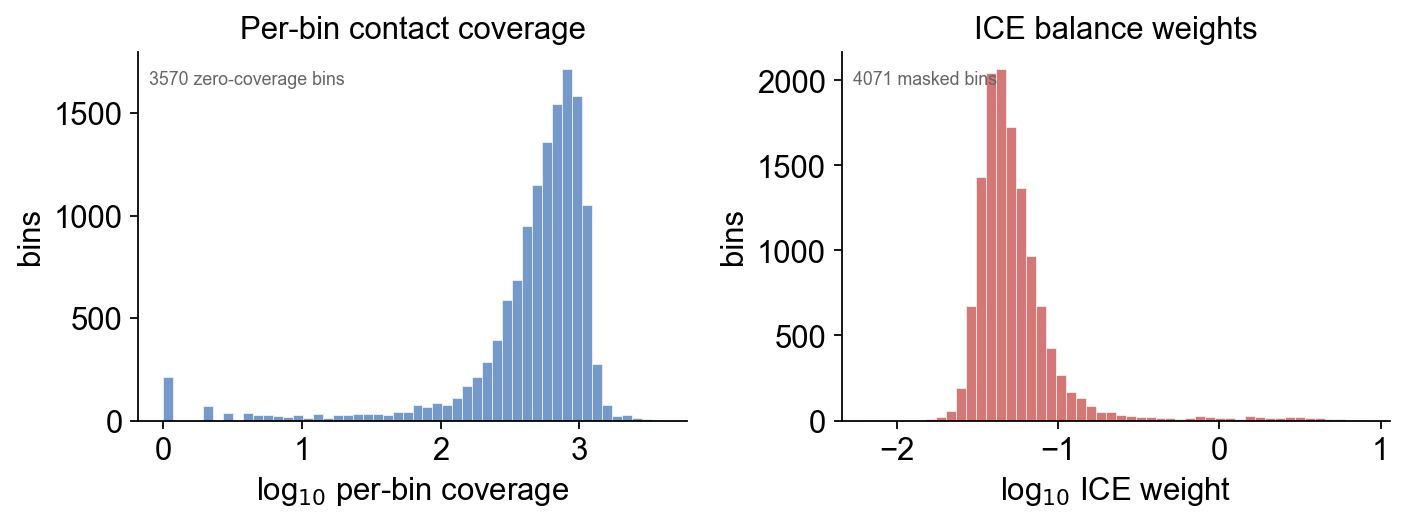
On the Drosophila S2 library: ~4 k bins are masked (no ICE weight), almost all of them on the Het / U / Uextra unmappable regions that dm3 pads with low-quality reference sequence. The rest of the 16.8 k bins retain balanced contacts.
6 · Summary + what’s next#
Phase 1 lands five epione.bulk.hic / epione.upstream entry points, all exercised above
against the Galaxy Drosophila dataset:
Call |
Role |
|---|---|
|
BAM → |
|
|
|
ICE-balance a |
|
log-scale region heatmap (chrom or interval) |
|
P(s) contact-decay curve |
|
per-bin coverage + ICE weight histograms |
Phase 2 (t_hic_analysis.ipynb) layers downstream analyses on top
of the same balanced .cool:
A / B compartments via eigendecomposition (
hicPCA-equivalent), with GC-content orientation.TAD boundaries via insulation score (
cooltools.insulation).Loop calling via dot-finder (
cooltools.dots).A composite “Hi-C triangle + TAD boundaries + ChIP-seq tracks” plot piped into the existing
epi.bulk.bigwig.plot_track_multi.