Upstream tutorial: ChIP-seq#
This notebook is an epione-native rewrite of the Galaxy Training Network TAL1 ChIP-seq tutorial. The goal is to start from raw ChIP and input FASTQ files, preprocess each replicate, merge replicates into experiment-level BAMs, generate browser-ready bigWig tracks, call peaks with an input control, and finally inspect the signal around a tutorial-relevant locus and around transcription start sites.
Unlike the previous upstream notebooks, this tutorial uses single-end ChIP-seq reads. That is important because it demonstrates why the new epione.upstream.* package is a better home for upstream logic than the old bulk-only wrappers: the same alignment layer now supports both paired-end and single-end sequencing inputs.
The teaching dataset comes from the Galaxy TAL1 lesson and includes both ChIP and matched input FASTQs. To keep the notebook executable in omicdev, we use a reduced chr4 mouse reference and reuse the TAL1 locus from the Galaxy-provided regions-of-interest BED file.
Scope note. This notebook covers the upstream part of a single-end ChIP-seq workflow:
FASTQ -> filtered BAM -> merged BAM -> bigWig -> input-controlled peaks -> visual QC. It does not cover motif discovery, differential binding, or replicate correlation plots.
Data provenance#
The example files are from the Galaxy Training Network TAL1 ChIP-seq tutorial and are hosted on Zenodo. We use the G1E ChIP and input replicates because they provide both raw FASTQ controls and a compact tutorial-scale signal structure.
Item |
Source |
How it is used here |
|---|---|---|
G1E TAL1 R1/R2 |
Galaxy GTN TAL1 tutorial |
Single-end ChIP replicates for the target factor. |
G1E input R1/R2 |
Galaxy GTN TAL1 tutorial |
Matched single-end control replicates used for MACS2 control subtraction. |
Regions of interest BED |
Galaxy GTN TAL1 tutorial |
Supplies the |
GRCm38 FASTA + Gencode annotation |
|
Used to derive a lightweight |
Learning goals#
By the end of the notebook, you should be able to answer five practical questions:
How does
epione.upstreamhandle single-end ChIP-seq inputs?Which preprocessing steps are replicate-specific, and which are experiment-level?
How do we call MACS2 peaks with a matched input control through
epione?How can we reuse a Galaxy tutorial locus inside
epi.bulk.bigwigvisualization?How can the same
epi.upstream.*API be transferred to real project ChIP-seq samples?
1. Check upstream tools#
The epione.upstream namespace keeps the alignment and file-conversion layer explicit. For this ChIP-seq tutorial, the critical external binaries are still the standard aligner and interval-processing tools, while bigWig writing and TSS aggregation are handled inside Python.
import epione as epi
epi.upstream.check_tools([
'bowtie2',
'samtools',
'bedtools',
'macs2',
])
✓ bowtie2 /scratch/users/steorra/env/omicdev/bin/bowtie2
✓ samtools /scratch/users/steorra/env/omicdev/bin/samtools
✓ bedtools /scratch/users/steorra/env/omicdev/bin/bedtools
✓ macs2 /scratch/users/steorra/env/omicdev/bin/macs2
{'bowtie2': '/scratch/users/steorra/env/omicdev/bin/bowtie2',
'samtools': '/scratch/users/steorra/env/omicdev/bin/samtools',
'bedtools': '/scratch/users/steorra/env/omicdev/bin/bedtools',
'macs2': '/scratch/users/steorra/env/omicdev/bin/macs2'}
2. Download the Galaxy ChIP and input FASTQs#
The TAL1 lesson ships both ChIP and matched input as single-end FASTQ replicates, which makes it more suitable than the previous toy ChIP example. We also download the Galaxy-provided regions-of-interest BED so that later visualization can reuse the Tal1 locus from the teaching material.
import os
import pathlib
import urllib.request
import warnings
import pandas as pd
import epione as epi
warnings.filterwarnings('ignore', message='Function .* is missing a docstring.*')
os.environ['LC_ALL'] = 'C'
os.environ['LC_CTYPE'] = 'C'
epi.pl.plot_set()
WORK = pathlib.Path.cwd()
DATA = WORK / 'galaxy_chipseq_demo'
FASTQ_DIR = DATA / 'fastq'
REF_DIR = DATA / 'ref'
OUT = DATA / 'result'
for p in [FASTQ_DIR, REF_DIR, OUT]:
p.mkdir(parents=True, exist_ok=True)
CHIP_URLS = {
'G1E_Tal1_R1.fastqsanger': 'https://zenodo.org/api/records/197100/files/G1E_Tal1_R1_downsampled_SRR492444.fastqsanger/content',
'G1E_Tal1_R2.fastqsanger': 'https://zenodo.org/api/records/197100/files/G1E_Tal1_R2_downsampled_SRR492445.fastqsanger/content',
'G1E_input_R1.fastqsanger': 'https://zenodo.org/api/records/197100/files/G1E_input_R1_downsampled_SRR507859.fastqsanger/content',
'G1E_input_R2.fastqsanger': 'https://zenodo.org/api/records/197100/files/G1E_input_R2_downsampled_SRR507860.fastqsanger/content',
'ChIPseq_regions_of_interest_v4.bed': 'https://zenodo.org/api/records/197100/files/ChIPseq_regions_of_interest_v4.bed/content',
}
└─ 🔬 Starting plot initialization...
├─ Apply Scanpy/matplotlib settings
├─ Custom font setup
├─ Suppress warnings
├─
___________ .__
\_ _____/_____ |__| ____ ____ ____
| __)_\____ \| |/ _ \ / \_/ __ \
| \ |_> > ( <_> ) | \ ___/
/_______ / __/|__|\____/|___| /\___ >
\/|__| \/ \/
├─ 🔖 Version: 0.0.1rc1 📚 Tutorials: https://epione.readthedocs.io/
└─ ✅ plot_set complete.
for name, url in CHIP_URLS.items():
out = FASTQ_DIR / name
if not out.exists():
print(f'downloading {name} ...')
urllib.request.urlretrieve(url, out)
else:
print(f'skip existing: {name}')
chip_reps = [
('Tal1_R1', FASTQ_DIR / 'G1E_Tal1_R1.fastqsanger'),
('Tal1_R2', FASTQ_DIR / 'G1E_Tal1_R2.fastqsanger'),
]
input_reps = [
('Input_R1', FASTQ_DIR / 'G1E_input_R1.fastqsanger'),
('Input_R2', FASTQ_DIR / 'G1E_input_R2.fastqsanger'),
]
regions_bed = FASTQ_DIR / 'ChIPseq_regions_of_interest_v4.bed'
pd.DataFrame({
'sample': [s for s, _ in chip_reps + input_reps],
'fastq': [str(fq) for _, fq in chip_reps + input_reps],
})
skip existing: G1E_Tal1_R1.fastqsanger
skip existing: G1E_Tal1_R2.fastqsanger
skip existing: G1E_input_R1.fastqsanger
skip existing: G1E_input_R2.fastqsanger
skip existing: ChIPseq_regions_of_interest_v4.bed
| sample | fastq | |
|---|---|---|
| 0 | Tal1_R1 | /scratch/users/steorra/analysis/omicverse_dev/... |
| 1 | Tal1_R2 | /scratch/users/steorra/analysis/omicverse_dev/... |
| 2 | Input_R1 | /scratch/users/steorra/analysis/omicverse_dev/... |
| 3 | Input_R2 | /scratch/users/steorra/analysis/omicverse_dev/... |
3. Prepare a chr4 mouse demo reference#
The Galaxy regions-of-interest file includes a Tal1 locus on chr4, so we use chr4 as the reduced executable reference. This keeps index construction small enough for a notebook run while preserving a locus that is explicitly part of the GTN teaching materials.
%%time
from pyfaidx import Fasta
MM10_FASTA = pathlib.Path(epi.upstream.fetch_genome_fasta('GRCm38'))
CHR4_FASTA = REF_DIR / 'chr4.fa'
CHR4_BT2_PREFIX = REF_DIR / 'chr4'
fa = Fasta(str(MM10_FASTA))
CHR4_SIZE = len(fa['chr4'])
if not CHR4_FASTA.exists():
with open(CHR4_FASTA, 'w') as fout:
fout.write('>chr4\n')
seq = str(fa['chr4'])
for i in range(0, len(seq), 60):
fout.write(seq[i:i+60] + '\n')
ref = epi.upstream.prepare_reference(
fasta=CHR4_FASTA,
aligner='bowtie2',
index_prefix=CHR4_BT2_PREFIX,
)
ref
CPU times: user 447 ms, sys: 90 ms, total: 537 ms
Wall time: 551 ms
{'fasta': '/scratch/users/steorra/analysis/omicverse_dev/epione/epione_guide/tutorials/bulk/galaxy_chipseq_demo/ref/chr4.fa',
'fai': '/scratch/users/steorra/analysis/omicverse_dev/epione/epione_guide/tutorials/bulk/galaxy_chipseq_demo/ref/chr4.fa.fai',
'chrom_sizes': '/scratch/users/steorra/analysis/omicverse_dev/epione/epione_guide/tutorials/bulk/galaxy_chipseq_demo/ref/chr4.chrom.sizes',
'ref_index': '/scratch/users/steorra/analysis/omicverse_dev/epione/epione_guide/tutorials/bulk/galaxy_chipseq_demo/ref/chr4'}
4. Process single-end ChIP and input replicates step by step#
The TAL1 lesson contains both ChIP FASTQs and matched input FASTQs, so we can express the complete single-end backbone explicitly for both signal and control.
For every replicate we run the same sequence of operations:
epi.upstream.bowtie2.align_fastq_to_bam(...)aligns the single-end reads and performs duplicate removal.epi.upstream.samtools.filter_bam(...)applies the mapping-quality and flag filters appropriate for single-end data.epi.upstream.samtools.index_bam(...)makes the filtered BAM directly reusable.epi.upstream.bigwig.bam_to_bigwig(...)exports the signal track used for browser-style inspection.
This makes the control logic obvious: input is not a special file type here, just another single-end alignment stream processed with the same core machinery.
%%time
chip_runs = {}
for sample_name, fq in chip_reps:
sample_dir = OUT / sample_name
sample_dir.mkdir(parents=True, exist_ok=True)
raw_bam = sample_dir / f'{sample_name}.raw.bam'
filt_bam = sample_dir / f'{sample_name}.filtered.bam'
bw_path = sample_dir / f'{sample_name}.rpkm.bw'
for stale in [raw_bam, filt_bam, pathlib.Path(str(filt_bam) + '.bai'), bw_path]:
if stale.exists():
stale.unlink()
epi.upstream.bowtie2.align_fastq_to_bam(
fq1=str(fq),
fq2=None,
out_bam=raw_bam,
ref_index=ref['ref_index'],
threads=8,
extra_args=['--very-sensitive'],
remove_duplicates=True,
)
epi.upstream.samtools.filter_bam(
raw_bam,
filt_bam,
mapq=30,
proper_pair=False,
drop_secondary_supp=True,
drop_duplicates=False,
drop_qcfail=False,
drop_unmapped=True,
drop_mate_unmapped=False,
threads=8,
)
epi.upstream.samtools.index_bam(filt_bam, threads=8)
epi.upstream.bigwig.bam_to_bigwig(
filt_bam,
bw_path,
bin_size=25,
normalize_using='RPKM',
threads=8,
)
raw_bam.unlink(missing_ok=True)
chip_runs[sample_name] = {'bam': str(filt_bam), 'bigwig': str(bw_path)}
input_runs = {}
for sample_name, fq in input_reps:
sample_dir = OUT / sample_name
sample_dir.mkdir(parents=True, exist_ok=True)
raw_bam = sample_dir / f'{sample_name}.raw.bam'
filt_bam = sample_dir / f'{sample_name}.filtered.bam'
bw_path = sample_dir / f'{sample_name}.rpkm.bw'
for stale in [raw_bam, filt_bam, pathlib.Path(str(filt_bam) + '.bai'), bw_path]:
if stale.exists():
stale.unlink()
epi.upstream.bowtie2.align_fastq_to_bam(
fq1=str(fq),
fq2=None,
out_bam=raw_bam,
ref_index=ref['ref_index'],
threads=8,
extra_args=['--very-sensitive'],
remove_duplicates=True,
)
epi.upstream.samtools.filter_bam(
raw_bam,
filt_bam,
mapq=30,
proper_pair=False,
drop_secondary_supp=True,
drop_duplicates=False,
drop_qcfail=False,
drop_unmapped=True,
drop_mate_unmapped=False,
threads=8,
)
epi.upstream.samtools.index_bam(filt_bam, threads=8)
epi.upstream.bigwig.bam_to_bigwig(
filt_bam,
bw_path,
bin_size=25,
normalize_using='RPKM',
threads=8,
)
raw_bam.unlink(missing_ok=True)
input_runs[sample_name] = {'bam': str(filt_bam), 'bigwig': str(bw_path)}
{
'chip': pd.DataFrame(chip_runs).T,
'input': pd.DataFrame(input_runs).T,
}
CPU times: user 2min 7s, sys: 3.41 s, total: 2min 11s
Wall time: 2min 41s
{'chip': bam \
Tal1_R1 /scratch/users/steorra/analysis/omicverse_dev/...
Tal1_R2 /scratch/users/steorra/analysis/omicverse_dev/...
bigwig
Tal1_R1 /scratch/users/steorra/analysis/omicverse_dev/...
Tal1_R2 /scratch/users/steorra/analysis/omicverse_dev/... ,
'input': bam \
Input_R1 /scratch/users/steorra/analysis/omicverse_dev/...
Input_R2 /scratch/users/steorra/analysis/omicverse_dev/...
bigwig
Input_R1 /scratch/users/steorra/analysis/omicverse_dev/...
Input_R2 /scratch/users/steorra/analysis/omicverse_dev/... }
5. Merge ChIP and input replicates#
The next transition is from replicate-level preprocessing to experiment-level summarisation. We merge the filtered ChIP replicates into one signal BAM, merge the filtered input replicates into one control BAM, and then export bigWigs for both.
This preserves the matched-control logic very transparently:
Alignment set |
Operation |
Result |
|---|---|---|
ChIP replicates |
|
Experiment-level signal BAM and bigWig. |
Input replicates |
|
Experiment-level control BAM and bigWig. |
The merged BAMs are the objects used by the downstream peak-calling step.
%%time
merged_dir = OUT / 'merged'
merged_dir.mkdir(parents=True, exist_ok=True)
for stale in [
merged_dir / 'Tal1.filtered.bam',
merged_dir / 'Tal1.filtered.bam.bai',
merged_dir / 'Tal1.rpkm.bw',
merged_dir / 'Input.filtered.bam',
merged_dir / 'Input.filtered.bam.bai',
merged_dir / 'Input.rpkm.bw',
]:
if stale.exists():
stale.unlink()
merged_chip_bam = epi.upstream.samtools.merge_bams(
[chip_runs['Tal1_R1']['bam'], chip_runs['Tal1_R2']['bam']],
merged_dir / 'Tal1.filtered.bam',
threads=8,
index=True,
)
merged_input_bam = epi.upstream.samtools.merge_bams(
[input_runs['Input_R1']['bam'], input_runs['Input_R2']['bam']],
merged_dir / 'Input.filtered.bam',
threads=8,
index=True,
)
merged_chip_bw = epi.upstream.bigwig.bam_to_bigwig(
merged_chip_bam,
merged_dir / 'Tal1.rpkm.bw',
bin_size=25,
normalize_using='RPKM',
threads=8,
)
merged_input_bw = epi.upstream.bigwig.bam_to_bigwig(
merged_input_bam,
merged_dir / 'Input.rpkm.bw',
bin_size=25,
normalize_using='RPKM',
threads=8,
)
{
'merged_chip_bam': merged_chip_bam,
'merged_input_bam': merged_input_bam,
'merged_chip_bigwig': merged_chip_bw,
'merged_input_bigwig': merged_input_bw,
}
CPU times: user 1min 3s, sys: 1.51 s, total: 1min 5s
Wall time: 1min 5s
{'merged_chip_bam': '/scratch/users/steorra/analysis/omicverse_dev/epione/epione_guide/tutorials/bulk/galaxy_chipseq_demo/result/merged/Tal1.filtered.bam',
'merged_input_bam': '/scratch/users/steorra/analysis/omicverse_dev/epione/epione_guide/tutorials/bulk/galaxy_chipseq_demo/result/merged/Input.filtered.bam',
'merged_chip_bigwig': '/scratch/users/steorra/analysis/omicverse_dev/epione/epione_guide/tutorials/bulk/galaxy_chipseq_demo/result/merged/Tal1.rpkm.bw',
'merged_input_bigwig': '/scratch/users/steorra/analysis/omicverse_dev/epione/epione_guide/tutorials/bulk/galaxy_chipseq_demo/result/merged/Input.rpkm.bw'}
merged_chip_bam='/scratch/users/steorra/analysis/omicverse_dev/epione/case/otx2/galaxy_chipseq_demo/result/merged/Tal1.filtered.bam'
merged_input_bam='/scratch/users/steorra/analysis/omicverse_dev/epione/case/otx2/galaxy_chipseq_demo/result/merged/Input.filtered.bam'
merged_chip_bigwig='/scratch/users/steorra/analysis/omicverse_dev/epione/case/otx2/galaxy_chipseq_demo/result/merged/Tal1.rpkm.bw'
merged_input_bigwig='/scratch/users/steorra/analysis/omicverse_dev/epione/case/otx2/galaxy_chipseq_demo/result/merged/Input.rpkm.bw'
6. Call peaks with a matched input control#
This is the key ChIP-specific statistical step. The merged TAL1 BAM supplies the signal, while the merged input BAM supplies the matched background model.
The function#
epi.upstream.macs2.call_peaks_macs2(...) wraps the standard MACS2 callpeak interface.
Key parameter |
What it controls |
|---|---|
|
Signal alignment used as the treatment set. |
|
Matched input alignment used as the control set. |
|
Treats the data as single-end reads. |
|
Leaves MACS2 model building enabled for this single-end ChIP example. |
Because the notebook uses a reduced chr4 reference, the goal is to reproduce the tutorial logic on a compact locus-focused dataset rather than generate a full-genome binding map.
%%time
merged_chip_bam = OUT / 'merged' / 'Tal1.filtered.bam'
merged_input_bam = OUT / 'merged' / 'Input.filtered.bam'
peak_paths = epi.upstream.macs2.call_peaks_macs2(
bam=merged_chip_bam,
control_bam=merged_input_bam,
out_dir=OUT / 'peaks',
name='G1E_Tal1',
genome_size=str(CHR4_SIZE),
format='BAM',
qvalue=0.01,
keep_dup='all',
call_summits=True,
nomodel=False,
)
peak_paths
CPU times: user 1.36 ms, sys: 1.01 ms, total: 2.38 ms
Wall time: 326 ms
{'narrowPeak': '/scratch/users/steorra/analysis/omicverse_dev/epione/epione_guide/tutorials/bulk/galaxy_chipseq_demo/result/peaks/G1E_Tal1_peaks.narrowPeak',
'summits': '/scratch/users/steorra/analysis/omicverse_dev/epione/epione_guide/tutorials/bulk/galaxy_chipseq_demo/result/peaks/G1E_Tal1_summits.bed',
'xls': '/scratch/users/steorra/analysis/omicverse_dev/epione/epione_guide/tutorials/bulk/galaxy_chipseq_demo/result/peaks/G1E_Tal1_peaks.xls'}
peak_paths={'narrowPeak': '/scratch/users/steorra/analysis/omicverse_dev/epione/case/otx2/galaxy_chipseq_demo/result/peaks/G1E_Tal1_peaks.narrowPeak',
'summits': '/scratch/users/steorra/analysis/omicverse_dev/epione/case/otx2/galaxy_chipseq_demo/result/peaks/G1E_Tal1_summits.bed',
'xls': '/scratch/users/steorra/analysis/omicverse_dev/epione/case/otx2/galaxy_chipseq_demo/result/peaks/G1E_Tal1_peaks.xls'}
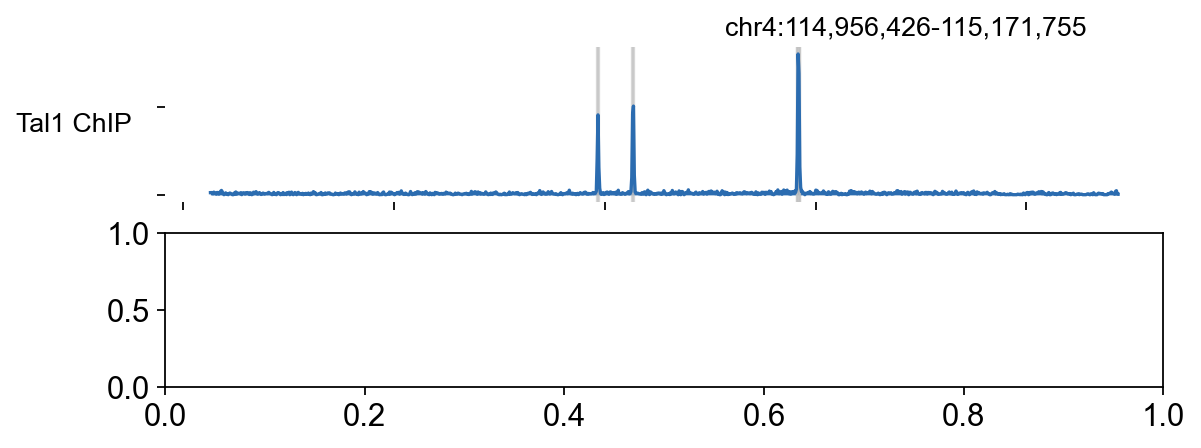
7. Visualise the Galaxy Tal1 locus with epi.bulk.bigwig#
The GTN tutorial ships a BED file of regions of interest that includes Tal1 on chr4. We reuse that teaching locus here, overlay the merged TAL1 and input bigWigs, and highlight any called peaks that overlap the window.
regions_df = pd.read_csv(regions_bed, sep=' ', header=None, names=['chrom', 'start', 'end', 'name'])
tal1_row = regions_df[regions_df['name'].str.lower() == 'tal1'].iloc[0]
TAL1_LOCUS = (str(tal1_row['chrom']), int(tal1_row['start']), int(tal1_row['end']))
peak_file = OUT / 'peaks' / 'G1E_Tal1_peaks.narrowPeak'
merged_chip_bw = str(OUT / 'merged' / 'Tal1.rpkm.bw')
merged_input_bw = str(OUT / 'merged' / 'Input.rpkm.bw')
peak_df = pd.read_csv(peak_file, sep=' ', header=None)
peak_df = peak_df[peak_df[0] == TAL1_LOCUS[0]].copy()
peak_df = peak_df[(peak_df[1] < TAL1_LOCUS[2]) & (peak_df[2] > TAL1_LOCUS[1])]
region_dict = {
f'peak_{i+1}': (int(r[1]), int(r[2]))
for i, r in peak_df.head(8).iterrows()
}
bw_obj = epi.bulk.bigwig({'Tal1 ChIP': merged_chip_bw, 'Input': merged_input_bw})
bw_obj.read()
color_dict = {'Tal1 ChIP': '#2B6CB0', 'Input': '#B0B0B0'}
bw_obj.plot_track(
chrom=TAL1_LOCUS[0],
chromstart=TAL1_LOCUS[1],
chromend=TAL1_LOCUS[2],
plot_names=['Tal1 ChIP', 'Input'],
figwidth=8,
figheight=2.8,
color_dict=color_dict,
bp_per_bin=200,
region_dict=region_dict,
)
└─ Load bigWig files
├─ Loading Tal1 ChIP...
└─ Loading Input...
(<Figure size 640x224 with 2 Axes>,
array([<Axes: ylabel='Tal1 ChIP'>, <Axes: >], dtype=object))
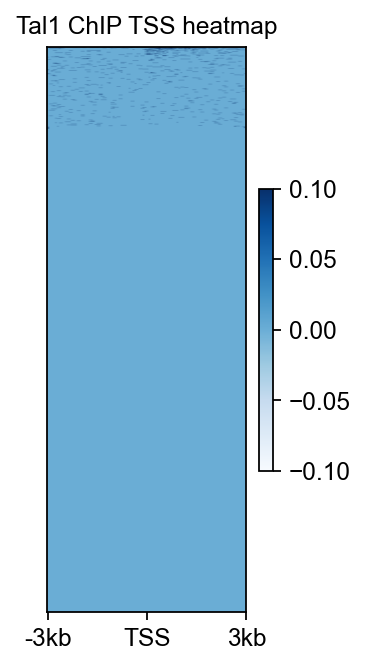
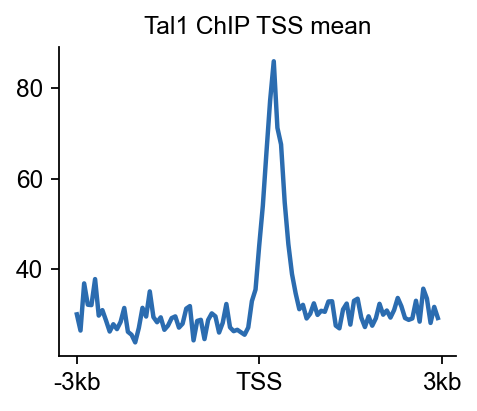
8. Compute a TSS heatmap for the merged ChIP signal#
The local browser view asks whether the Tal1 locus looks sensible. The TSS-centred heatmap asks a broader question: when the merged ChIP signal is summarized around annotated promoters on chr4, does it show structured enrichment rather than flat background?
To keep the annotation consistent with the reduced reference, we derive a transcript-only chr4 GTF from the matching mouse GENCODE gff3 file through epi.utils.convert_gff_to_gtf(...).
GENCODE_GFF3 = pathlib.Path(epi.upstream.fetch_genome_annotation('GRCm38'))
CHR4_GTF = REF_DIR / 'gencode_vM25_chr4.transcripts.gtf'
CHR4_GTF = pathlib.Path(
epi.utils.convert_gff_to_gtf(
GENCODE_GFF3,
CHR4_GTF,
feature_whitelist=['transcript'],
seqname_whitelist=['chr4'],
)
)
with open(CHR4_GTF) as fin:
transcript_count = sum(1 for _ in fin)
print(CHR4_GTF, transcript_count)
└─ Converting GFF to GTF: /tmp/snapatac2/gencode_vM25_GRCm38.gff3.gz -> /scratch/users/steorra/analysis/omicverse_dev/epione/epione_guide/tutorials/bulk/galaxy_chipseq_demo/ref/gencode_vM25_chr4.transcripts.gtf
└─ Wrote 7035 GTF records
/scratch/users/steorra/analysis/omicverse_dev/epione/epione_guide/tutorials/bulk/galaxy_chipseq_demo/ref/gencode_vM25_chr4.transcripts.gtf 7035
merged_chip_bw = str(OUT / 'merged' / 'Tal1.rpkm.bw')
chip_tss = epi.bulk.bigwig({'Tal1 ChIP': merged_chip_bw})
chip_tss.read()
chip_tss.load_gtf(str(CHR4_GTF))
chip_tss.compute_matrix('Tal1 ChIP', nbins=100, upstream=3000, downstream=3000, n_jobs=1)
fig, ax = chip_tss.plot_matrix(
bw_name='Tal1 ChIP',
bw_type='TSS',
figsize=(2.4, 4.2),
cmap='Blues',
vmax='auto',
vmin='auto',
fontsize=11,
title='Tal1 ChIP TSS heatmap',
)
chip_tss.plot_matrix_line(
bw_name='Tal1 ChIP',
bw_type='TSS',
figsize=(3.2, 2.6),
color='#2B6CB0',
fontsize=11,
title='Tal1 ChIP TSS mean',
)
└─ Load bigWig files
└─ Loading Tal1 ChIP...
└─ Load GTF file
├─ Reading GTF...
└─ Reading GTF file from /scratch/users/steorra/analysis/omicverse_dev/epione/epione_guide/tutorials/bulk/galaxy_chipseq_demo/ref/gencode_vM25_chr4.transcripts.gtf...
└─ GTF file read successfully
└─ GTF loaded
└─ Compute matrix: Tal1 ChIP
├─ Prepare features
├─ Build matrices
└─ Finalize
└─ Tal1 ChIP matrix finished
└─ Tal1 ChIP tss matrix in bw_tss_scores_dict[Tal1 ChIP]
└─ Tal1 ChIP tes matrix in bw_tes_scores_dict[Tal1 ChIP]
└─ Tal1 ChIP body matrix in bw_body_scores_dict[Tal1 ChIP]
(<Figure size 256x208 with 1 Axes>,
<Axes: title={'center': 'Tal1 ChIP TSS mean'}>)
9. OTX2 project template#
The final cell shows the project handoff point using the same explicit ChIP/input preprocessing structure as the tutorial itself. Replace the tutorial FASTQs and the reduced chr4 reference with your real project data, but keep the same step-by-step sequence of epi.upstream.bowtie2, epi.upstream.samtools, epi.upstream.bigwig, and epi.upstream.macs2 calls.
# chip_rep1_dir = WORK / 'otx2_chip_upstream' / 'chip_rep1'
# input_rep1_dir = WORK / 'otx2_chip_upstream' / 'input_rep1'
# chip_rep1_dir.mkdir(parents=True, exist_ok=True)
# input_rep1_dir.mkdir(parents=True, exist_ok=True)
#
# chip_rep1_raw = chip_rep1_dir / 'chip_rep1.raw.bam'
# chip_rep1_bam = chip_rep1_dir / 'chip_rep1.filtered.bam'
# chip_rep1_bw = chip_rep1_dir / 'chip_rep1.rpkm.bw'
# input_rep1_raw = input_rep1_dir / 'input_rep1.raw.bam'
# input_rep1_bam = input_rep1_dir / 'input_rep1.filtered.bam'
# input_rep1_bw = input_rep1_dir / 'input_rep1.rpkm.bw'
#
# epi.upstream.bowtie2.align_fastq_to_bam(
# fq1='/path/to/chip_rep1.fastq.gz',
# fq2=None,
# out_bam=chip_rep1_raw,
# ref_index='/path/to/full/genome/index/prefix',
# threads=8,
# extra_args=['--very-sensitive'],
# remove_duplicates=True,
# )
# epi.upstream.samtools.filter_bam(chip_rep1_raw, chip_rep1_bam, mapq=30, proper_pair=False, drop_secondary_supp=True, drop_unmapped=True, drop_mate_unmapped=False, threads=8)
# epi.upstream.samtools.index_bam(chip_rep1_bam, threads=8)
# epi.upstream.bigwig.bam_to_bigwig(chip_rep1_bam, chip_rep1_bw, bin_size=25, normalize_using='RPKM', threads=8)
#
# epi.upstream.bowtie2.align_fastq_to_bam(
# fq1='/path/to/input_rep1.fastq.gz',
# fq2=None,
# out_bam=input_rep1_raw,
# ref_index='/path/to/full/genome/index/prefix',
# threads=8,
# extra_args=['--very-sensitive'],
# remove_duplicates=True,
# )
# epi.upstream.samtools.filter_bam(input_rep1_raw, input_rep1_bam, mapq=30, proper_pair=False, drop_secondary_supp=True, drop_unmapped=True, drop_mate_unmapped=False, threads=8)
# epi.upstream.samtools.index_bam(input_rep1_bam, threads=8)
# epi.upstream.bigwig.bam_to_bigwig(input_rep1_bam, input_rep1_bw, bin_size=25, normalize_using='RPKM', threads=8)
#
# merged_chip_bam = epi.upstream.samtools.merge_bams([chip_rep1_bam], WORK / 'otx2_chip_upstream' / 'chip.bam', threads=8)
# merged_input_bam = epi.upstream.samtools.merge_bams([input_rep1_bam], WORK / 'otx2_chip_upstream' / 'input.bam', threads=8)
# peak_paths = epi.upstream.macs2.call_peaks_macs2(
# bam=merged_chip_bam,
# control_bam=merged_input_bam,
# out_dir=WORK / 'otx2_chip_upstream' / 'peaks',
# name='chip_merged',
# genome_size='mm',
# format='BAM',
# qvalue=0.01,
# keep_dup='all',
# call_summits=True,
# nomodel=False,
# )