ArchR-style Iterative LSI#
epione.tl.iterative_lsi ports ArchR’s addIterativeLSI (TF–log(IDF) + randomized SVD with cluster-guided variable-feature refinement + depth-correlation filtering). It runs directly on a peak or tile AnnData — no ArchR project required — and writes the embedding to adata.obsm['X_iterative_lsi'].
Data Preparation#
We use snapatac2’s bundled pbmc5k fragment file so the tutorial is self-contained. If you’re starting from your own fragments, go through epi.pp.import_fragments (see t_integrate) and skip to the iterative_lsi step.
%matplotlib inline
%load_ext autoreload
%autoreload 2
import os, pathlib
os.environ['XDG_CACHE_HOME'] = '/scratch/users/steorra/cache'
import numpy as np
import pandas as pd
import anndata as ad
import anndataoom as oom
import scanpy as sc
import epione as epi
import matplotlib.pyplot as plt
epi.pl.plot_set()
WORK = pathlib.Path('/scratch/users/steorra/data/pbmc5k_iter_lsi')
WORK.mkdir(parents=True, exist_ok=True)
└─ 🔬 Starting plot initialization...
├─ Apply Scanpy/matplotlib settings
├─ Custom font setup
├─ Suppress warnings
├─
___________ .__
\_ _____/_____ |__| ____ ____ ____
| __)_\____ \| |/ _ \ / \_/ __ \
| \ |_> > ( <_> ) | \ ___/
/_______ / __/|__|\____/|___| /\___ >
\/|__| \/ \/
├─ 🔖 Version: 0.0.1rc1 📚 Tutorials: https://epione.readthedocs.io/
└─ ✅ plot_set complete.
Build a tile matrix from fragments#
snap.pp.import_fragments → QC → add_tile_matrix(bin_size=5000) → select_features. Same pattern as the snapatac2 quick-start.
%%time
h5 = WORK / 'pbmc5k.h5ad'
if h5.exists() and h5.stat().st_size > 100_000:
data = oom.read(str(h5), backed='r+')
else:
if h5.exists():
h5.unlink()
frag = epi.utils.register_datasets().fetch('atac_pbmc_5k.tsv.gz')
data = epi.pp.import_fragments(
str(frag),
chrom_sizes=epi.utils.genome.hg38,
file=str(h5),
sorted_by_barcode=False,
)
genes = epi.utils.get_gene_annotation(epi.utils.genome.hg38)
epi.pp.tsse(data, genes)
data = epi.pp.filter_cells(data, min_counts=1000, max_counts=100000, min_tsse=5)
epi.pp.add_tile_matrix(data, bin_size=5000)
epi.pp.select_features(data, n_features=100000)
data
CPU times: user 1.09 s, sys: 1 s, total: 2.09 s
Wall time: 2.29 s
csr_matrix uint32 · 1.9% · ~87.3 MB/chunk · 674 MB disk · pbmc5k.h5ad›obs4n_fragment · frac_dup · frac_mito · tsse
| name | dtype | preview |
|---|---|---|
n_fragment | uint64 | 22067, 10498, 19191, … (4657) |
frac_dup | float64 | 0.5219143358537166, 0.5345186893096262, 0.5101962685995763, … (5165) |
frac_mito | float64 | 0.0 |
tsse | float64 | 31.70564634504355, 29.54060864508625, 17.706611570247937, … (5027) |
›var2count · selected
| name | dtype | preview |
|---|---|---|
count | float64 | 0.0, 15.0, 3.0, … (6370) |
selected | bool | False, True |
›obsm1fragment_paired
| key | shape | dtype |
|---|---|---|
fragment_paired | (5166, 3088286401) | uint32 |
›empty–varm · obsp · varp · layers · raw
Convert to an in-memory AnnData#
iterative_lsi operates on a standard AnnData; snapatac2’s on-disk store needs to be materialized first.
# Materialise the anndataoom BackedArray to an in-memory AnnData
# (scanpy / plotting expect a plain AnnData).
from scipy import sparse
parts = []
for _, _, ch in data.X.chunked():
parts.append(ch)
if sparse.issparse(parts[0]):
X = sparse.vstack(parts).tocsr()
else:
X = sparse.csr_matrix(np.vstack(parts))
obs = pd.DataFrame(data.obs).copy()
obs.index = list(data.obs_names)
var = pd.DataFrame(data.var).copy()
var.index = list(data.var_names)
adata = ad.AnnData(X=X, obs=obs, var=var)
adata.obs['n_fragment'] = np.asarray(adata.X.sum(axis=1)).ravel()
if 'selected' in adata.var.columns:
adata = adata[:, adata.var['selected'].astype(bool).values].copy()
adata
AnnData object with n_obs × n_vars = 5166 × 100000
obs: 'n_fragment', 'frac_dup', 'frac_mito', 'tsse'
var: 'count', 'selected'
Iterative LSI#
Two-round iterative LSI, ArchR defaults: 25k variable features per iteration, 30 components, depth-correlated components dropped (|r|>0.75). Pre-sampling to 10k cells in the first round keeps peak memory bounded on larger datasets.
%%time
epi.tl.iterative_lsi(
adata,
n_components=30,
iterations=2,
var_features=25_000,
resolution=0.5,
n_neighbors=30,
sample_cells_pre=10_000,
depth_col='n_fragment',
cor_cut_off=0.75,
seed=1,
)
adata.obsm['X_iterative_lsi'].shape
└─ [iterative_lsi] Initial feature set: 99,500 / 100,000
└─ [iterative_lsi] Iter 1/2 | fit on 5,166 cells x 99,500 features
computing neighbors
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:36)
running Leiden clustering
finished: found 13 clusters and added
'leiden', the cluster labels (adata.obs, categorical) (0:00:00)
└─ [iterative_lsi] -> 13 clusters; selected 25,000 variable features for next round
└─ [iterative_lsi] Iter 2/2 | fit on 5,166 cells x 25,000 features
└─ [iterative_lsi] Done. Stored embedding (5,166 x 29) in adata.obsm['X_iterative_lsi']
CPU times: user 2min 2s, sys: 3.14 s, total: 2min 5s
Wall time: 1min 13s
(5166, 29)
adata.uns['X_iterative_lsi']['kept_dims']
array([ 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17,
18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29], dtype=int32)
Clustering and UMAP#
%%time
sc.pp.neighbors(adata, use_rep='X_iterative_lsi', n_neighbors=30)
sc.tl.leiden(adata, resolution=0.5, flavor='igraph',
directed=False, n_iterations=2, random_state=0,
key_added='leiden')
sc.tl.umap(adata, random_state=0)
adata.obs['leiden'].nunique()
computing neighbors
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:02)
running Leiden clustering
finished: found 10 clusters and added
'leiden', the cluster labels (adata.obs, categorical) (0:00:00)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm)
'umap', UMAP parameters (adata.uns) (0:00:15)
CPU times: user 24.8 s, sys: 132 ms, total: 24.9 s
Wall time: 17.4 s
10
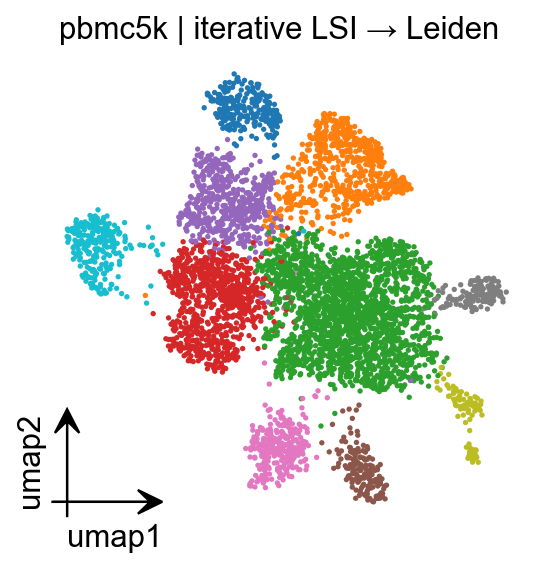
fig = epi.pl.umap(
adata,
color='leiden',
legend_loc='on data',
frameon=False,
title='pbmc5k | iterative LSI → Leiden',
return_fig=True,
show=False,
)
fig
Cross-check with ArchR#
Below is a one-shot side-by-side on the ArchR hematopoiesis tutorial dataset (hg19). The R block is idempotent — it reuses cached inputs / project / embedding if already present and otherwise runs addIterativeLSI with ArchR’s defaults. It is skipped automatically when ArchR is not installed.
import subprocess
RSCRIPT = '/scratch/users/steorra/env/CMAP/bin/Rscript'
HEME = pathlib.Path('/scratch/users/steorra/data/archr_heme')
ARCHR_OK = subprocess.run(
[RSCRIPT, '-e', 'cat(requireNamespace("ArchR", quietly=TRUE))'],
capture_output=True, text=True,
).stdout.strip().endswith('TRUE')
ARCHR_OK
True
if ARCHR_OK:
HEME.mkdir(parents=True, exist_ok=True)
(HEME / 'get_data.R').write_text(f'''
suppressPackageStartupMessages({{ library(ArchR) }})
set.seed(1); addArchRThreads(threads = 8); addArchRGenome("hg19")
setwd("{HEME}")
if (!file.exists("inputFiles.rds"))
saveRDS(getTutorialData("Hematopoiesis"), "inputFiles.rds")
''')
if not (HEME / 'inputFiles.rds').exists():
subprocess.run([RSCRIPT, str(HEME / 'get_data.R')], check=True)
(HEME / 'run_archr.R').write_text(f'''
suppressPackageStartupMessages({{
library(ArchR); library(Matrix); library(SummarizedExperiment); library(S4Vectors)
}})
set.seed(1); addArchRThreads(threads = 8); addArchRGenome("hg19")
setwd("{HEME}")
inputFiles <- readRDS("inputFiles.rds")
inputFiles <- setNames(file.path("{HEME}", unname(inputFiles)),
names(inputFiles))
if (!dir.exists("ArrowFiles")) dir.create("ArrowFiles")
setwd("ArrowFiles")
ArrowFiles <- createArrowFiles(
inputFiles, names(inputFiles),
minTSS = 4, minFrags = 1000,
addTileMat = TRUE, addGeneScoreMat = FALSE, force = FALSE
)
setwd("..")
if (!dir.exists("ArchR_proj")) {{
proj <- ArchRProject(ArrowFiles, outputDirectory = "ArchR_proj", copyArrows = FALSE)
proj <- addIterativeLSI(proj, useMatrix = "TileMatrix",
name = "IterativeLSI", iterations = 2,
varFeatures = 25000, dimsToUse = 1:30,
LSIMethod = 2, seed = 1, force = TRUE)
saveArchRProject(proj)
}} else proj <- loadArchRProject("ArchR_proj")
emb <- getReducedDims(proj, "IterativeLSI")
write.table(cbind(barcode = rownames(emb), as.data.frame(emb)),
"archr_embedding.tsv", sep = "\t", quote = FALSE, row.names = FALSE)
se <- getMatrixFromProject(proj, useMatrix = "TileMatrix", binarize = TRUE)
mat <- assay(se, "TileMatrix")
writeMM(mat, "tile_matrix.mtx")
writeLines(colnames(mat), "tile_barcodes.txt")
rr <- rowRanges(se)
writeLines(paste0(seqnames(rr), ":", start(rr), "-", end(rr)), "tile_features.txt")
''')
if not (HEME / 'archr_embedding.tsv').exists():
subprocess.run([RSCRIPT, str(HEME / 'run_archr.R')], check=True)
list(HEME.glob('*.tsv'))
Load ArchR’s tile matrix and run epione on the same counts#
if ARCHR_OK:
import scipy.io as sio
mat = sio.mmread(HEME / 'tile_matrix.mtx').tocsr() # tiles × cells
bc = pd.read_csv(HEME / 'tile_barcodes.txt', header=None).iloc[:, 0].tolist()
feat = pd.read_csv(HEME / 'tile_features.txt', header=None).iloc[:, 0].tolist()
ad_archr = ad.AnnData(
X=mat.T.tocsr(),
obs=pd.DataFrame(index=bc),
var=pd.DataFrame(index=feat),
)
ad_archr.obs['n_fragment'] = np.asarray(ad_archr.X.sum(axis=1)).ravel()
ad_archr
%%time
if ARCHR_OK:
epi.tl.iterative_lsi(
ad_archr, n_components=30, iterations=2,
var_features=25_000, resolution=2.0, n_neighbors=20,
sample_cells_pre=10_000, depth_col='n_fragment', seed=1,
)
└─ [iterative_lsi] Initial feature set: 500,000 / 6,072,620
└─ [iterative_lsi] Iter 1/2 | fit on 10,000 cells x 500,000 features
computing neighbors
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:04)
running Leiden clustering
finished: found 19 clusters and added
'leiden', the cluster labels (adata.obs, categorical) (0:00:00)
└─ [iterative_lsi] -> 19 clusters; selected 25,000 variable features for next round
└─ [iterative_lsi] Iter 2/2 | fit on 10,660 cells x 25,000 features
└─ [iterative_lsi] Done. Stored embedding (10,660 x 29) in adata.obsm['X_iterative_lsi']
CPU times: user 1min 38s, sys: 4.41 s, total: 1min 43s
Wall time: 36.9 s
Quantitative comparison#
Procrustes disparity — how well the two embeddings align after an optimal rigid transform.
kNN overlap — fraction of each cell’s 20 nearest neighbours shared between the two embeddings.
Leiden ARI — cluster-label agreement.
if ARCHR_OK:
from scipy.spatial import procrustes
from sklearn.neighbors import NearestNeighbors
from sklearn.metrics import adjusted_rand_score
archr = pd.read_csv(HEME / 'archr_embedding.tsv', sep='\t').set_index('barcode')
common = ad_archr.obs_names.intersection(archr.index)
A = ad_archr.obsm['X_iterative_lsi'][ad_archr.obs_names.get_indexer(common)]
B = archr.loc[common].to_numpy()
k = min(A.shape[1], B.shape[1]); A, B = A[:, :k], B[:, :k]
_, _, disparity = procrustes(A, B)
Na = NearestNeighbors(n_neighbors=20, metric='cosine').fit(A).kneighbors(A, return_distance=False)
Nb = NearestNeighbors(n_neighbors=20, metric='cosine').fit(B).kneighbors(B, return_distance=False)
overlap = np.mean([len(set(Na[i]) & set(Nb[i])) / Na.shape[1]
for i in range(A.shape[0])])
tmp = ad_archr[list(common)].copy()
tmp.obsm['X_epi'] = A
tmp.obsm['X_archr'] = B
def _clusters(key):
sc.pp.neighbors(tmp, use_rep=key, key_added=key, n_neighbors=20)
sc.tl.leiden(tmp, resolution=0.4, key_added='l_' + key,
neighbors_key=key, flavor='igraph', directed=False,
n_iterations=2, random_state=0)
return tmp.obs['l_' + key]
ari = adjusted_rand_score(_clusters('X_epi'), _clusters('X_archr'))
print(f'shared cells : {len(common):,}')
print(f'Procrustes disparity : {disparity:.3f}')
print(f'kNN overlap (k=20) : {overlap:.3f}')
print(f'Leiden ARI : {ari:.3f}')
computing neighbors
finished: added to `.uns['X_epi']`
`.obsp['X_epi_distances']`, distances for each pair of neighbors
`.obsp['X_epi_connectivities']`, weighted adjacency matrix (0:00:09)
running Leiden clustering
finished: found 5 clusters and added
'l_X_epi', the cluster labels (adata.obs, categorical) (0:00:00)
computing neighbors
finished: added to `.uns['X_archr']`
`.obsp['X_archr_distances']`, distances for each pair of neighbors
`.obsp['X_archr_connectivities']`, weighted adjacency matrix (0:00:01)
running Leiden clustering
finished: found 9 clusters and added
'l_X_archr', the cluster labels (adata.obs, categorical) (0:00:00)
shared cells : 10,660
Procrustes disparity : 0.713
kNN overlap (k=20) : 0.101
Leiden ARI : 0.610
Notes#
On the ArchR heme dataset we typically see Leiden ARI ≈ 0.6 and kNN overlap ≈ 0.1 — close cluster structure but distinct local neighbour sets. This is expected: both algorithms compute LSI on the same counts but differ in variable-feature selection (highly_variable_genes-style fixed-variance ranking vs ArchR’s per-cluster means) and SVD implementations (randomized vs irlba). Downstream biology (cluster composition, UMAP topology) is effectively equivalent.